
研究背景
氧还原反应(ORR)是燃料电池、金属空气电池等先进能源存储技术中的关键反应。目前,铂基金属催化剂因其优异的活性而被广泛使用,但高昂的价格、较差的耐久性和有限的自然丰度限制了它们的大规模应用。因此,寻找更经济、高效的非贵金属催化剂,以减少对贵金属的依赖,成为了推动先进能源存储技术发展的重要任务。非贵金属电催化剂,包括碳基材料、金属氧化物、尖晶石和钙钛矿等,已被研究用于ORR。其中,碳材料因其良好的稳定性、高导电性、大比表面积、简单的制备方法和丰富的可用性而成为有前途的候选材料。然而,纯碳基材料与含氧中间体之间的固有弱相互作用通常导致其ORR活性和选择性较差。杂原子掺杂是一种提高碳基催化剂性能的有前景的方法,因为它独特的电子密度分布以及优异的ORR活性。已有研究表明,杂原子(如氮、硼、磷、硫)掺杂的碳基催化剂具有良好的ORR催化活性。硅作为一种非金属杂原子,其电负性仅为1.90,远小于碳和氧原子的电负性。通过将硅掺杂到碳基材料中,电负性的差异导致硅失去电子并带正电,从而增强了对电子氧的吸附。这创造了额外的活性位点,有望显著提高ORR性能。
成果简介
在这项研究中,研究人员成功合成了二维硅掺杂石墨烯类材料(Si-GLC),其特点是含有“C-O-Si”键,通过原位掺杂方法实现。由于硅的电负性远小于碳和氧,形成“C-O-Si”键导致硅失去大量电子并带正电,这增加了对电子氧的吸附,从而提高了氧还原反应的活性。通过密度泛函理论计算,研究人员发现Si-GLC对氧分子的吸附能远低于石墨烯类碳(GLC),表明硅掺杂增强了石墨烯类材料对氧分子的吸附,这对提高氧还原反应的性能至关重要。Si-GLC在0.1 M KOH溶液中显示出优异的催化活性,半波电位为0.80 V(相对于RHE),扩散限制电流密度为5.81 mA·cm-2,展现出良好的稳定性和对甲醇交叉效应的耐受性。原位掺杂创造的“C-O-Si”键为设计高效的非金属基ORR催化剂提供了新策略。
图文导读
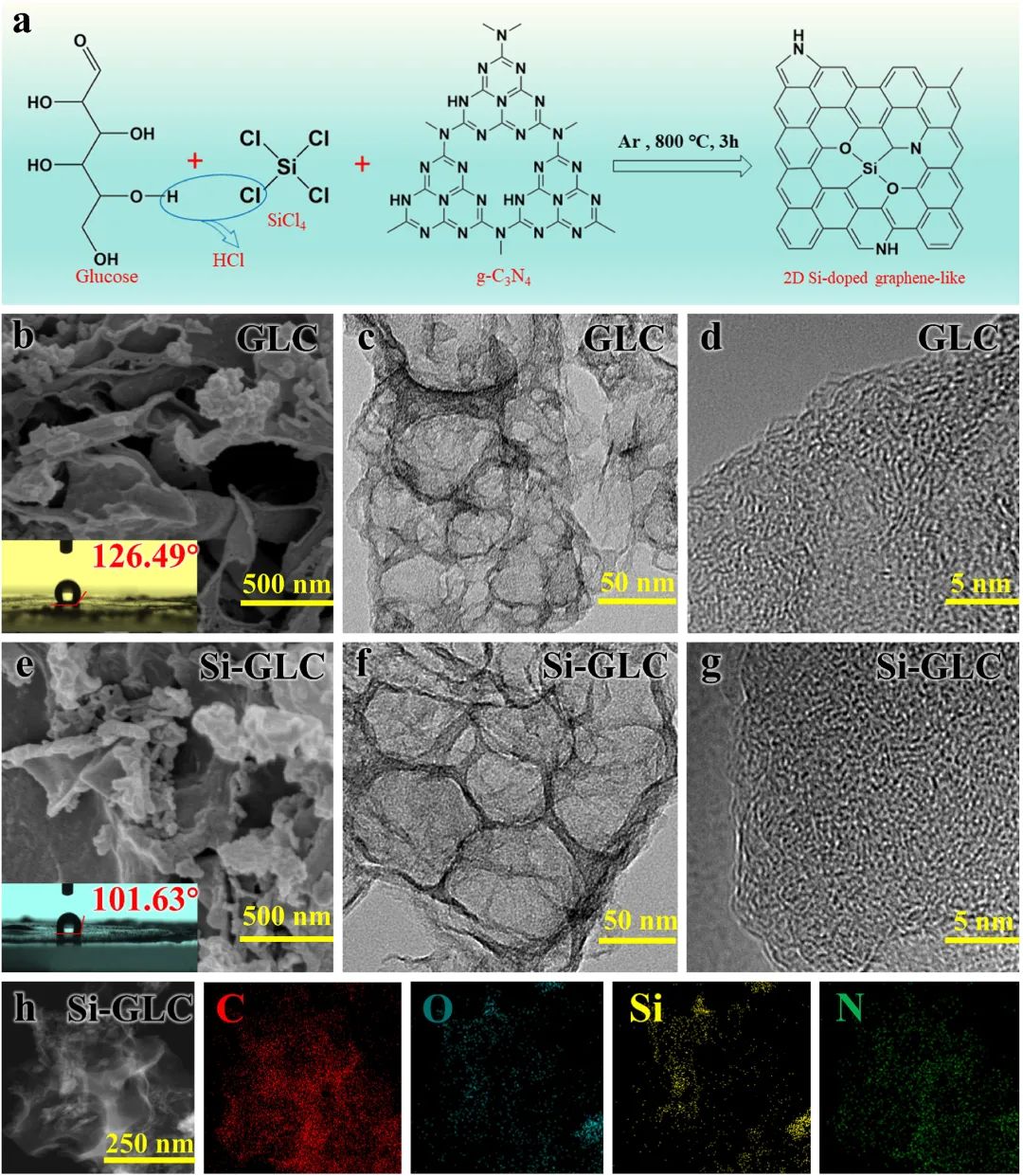

图1:展示了2D硅掺杂石墨烯类材料的制备示意图;GLC和Si-GLC的扫描/透射电子显微镜图像,显示了样品的纳米片结构和微孔结构;Si-GLC的TEM图像和相应的EDS映射显示了硅的成功掺杂和均匀分布。
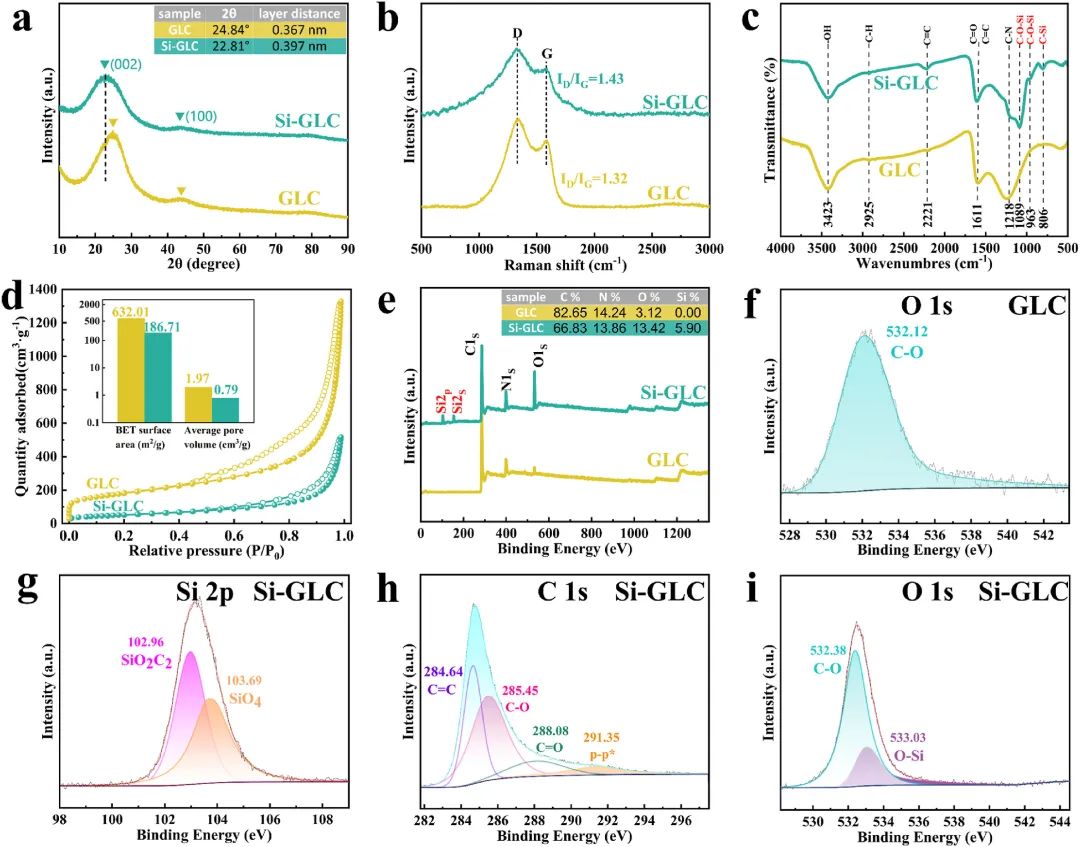
图2:XRD、拉曼、FT-IR、N2吸附-脱附等温线图展示了GLC和Si-GLC的晶体结构、晶体学信息、化学键和官能团;XPS全谱和O 1s、C 1s、Si 2p的峰进一步证实了硅的成功掺杂和特定价态。
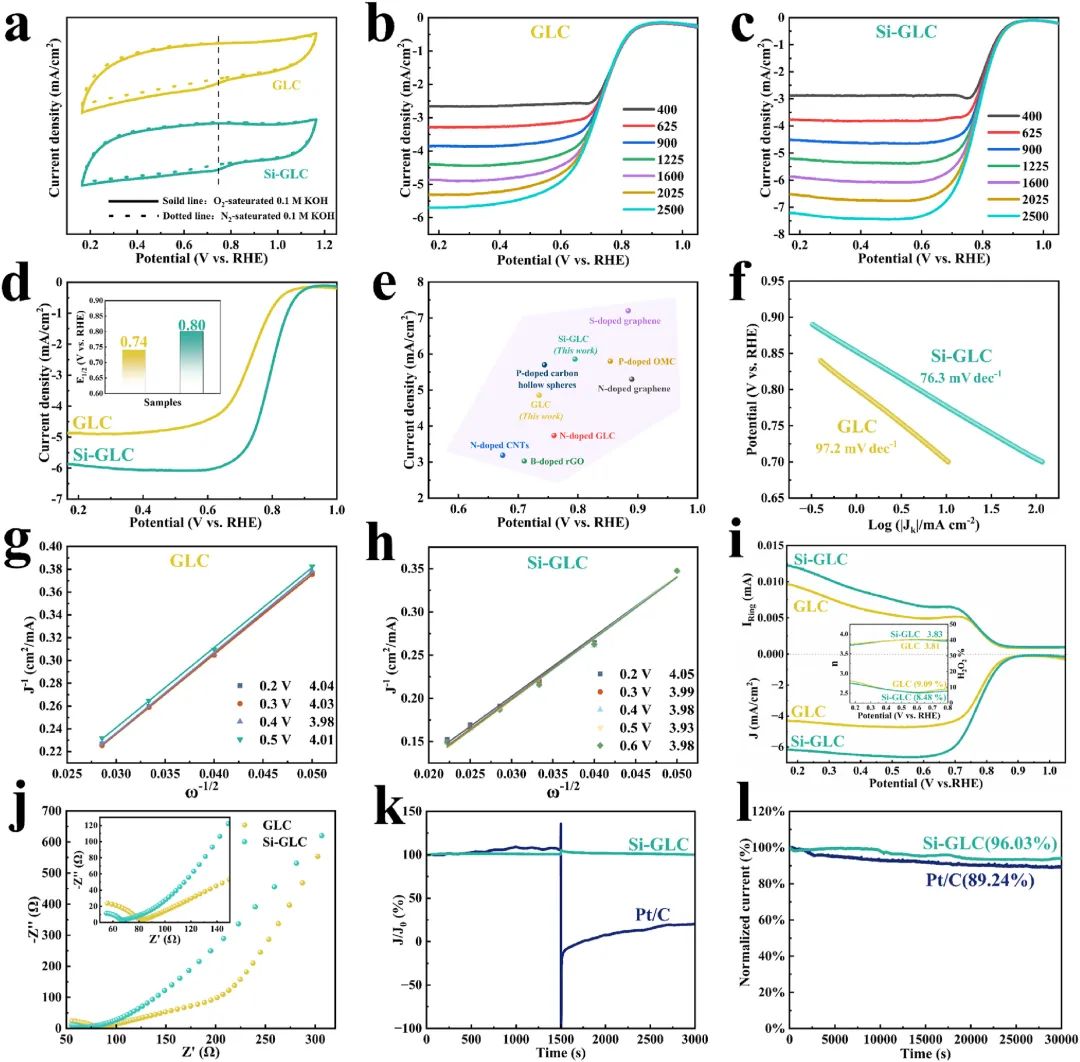
图3:CV曲线、LSV曲线、扩散限制电流密度和半波电位的比较、Tafel曲线、K-L曲线、RRDE伏安图和EIS曲线展示了GLC和Si-GLC的电化学性能,包括ORR活性、电子转移数、H2O2产生百分比和催化剂稳定性。
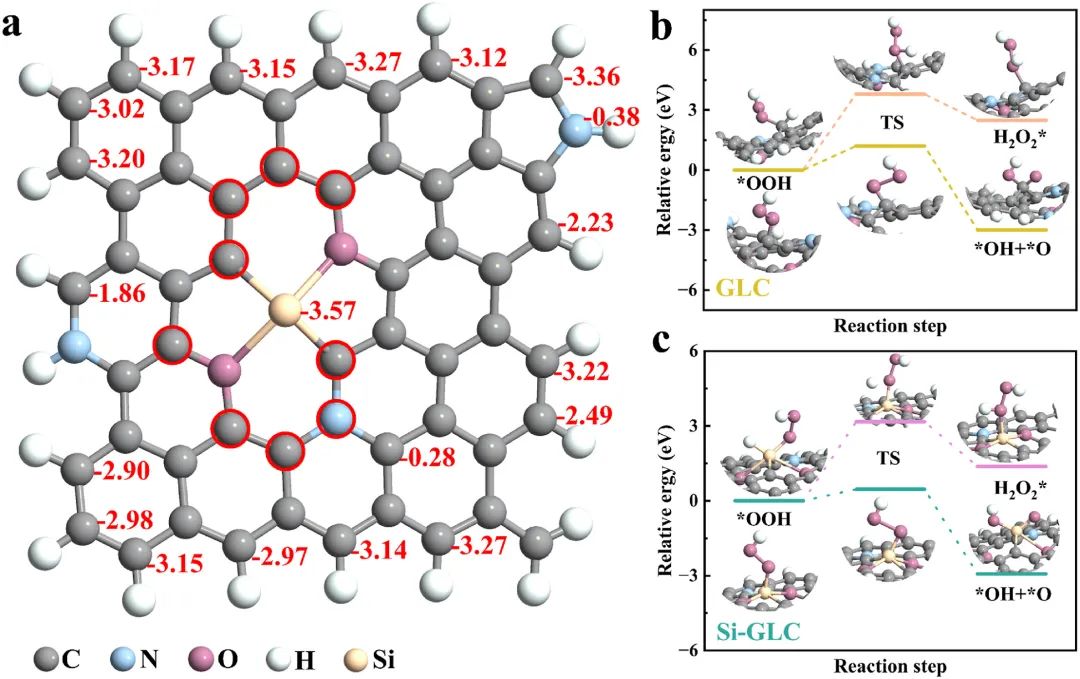
图4:展示了Si-GLC上不同位点的氧吸附能;OOH解离形成O和*OH的能量障碍,以及H添加形成HOOH的过程。
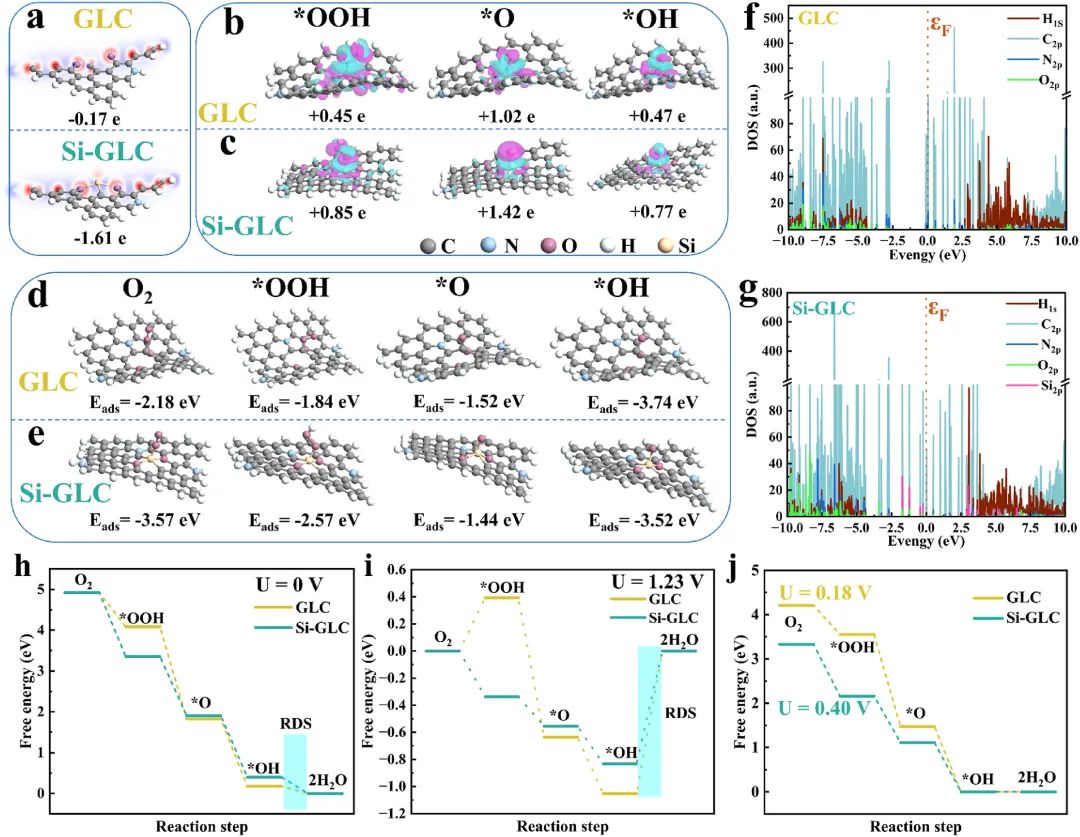
图5:电荷密度差异图、Bader电荷转移数、O2分子、*OOH、O和OH中间体在GLC和Si-GLC模型上的优化配置和吸附能;PDOS计算和在不同电极电位下四电子ORR的吉布斯自由能剖面图。
小结
研究人员通过原位掺杂方法成功合成了具有“C-O-Si”键的二维硅掺杂石墨烯类材料(Si-GLC),并展示了其在氧还原反应中的高效催化性能。密度泛函理论计算揭示了硅掺杂对碳材料ORR活性的电子结构调控作用。硅掺杂碳材料中带正电的活性位点增强了对电子氧的吸附,显著提高了ORR的催化活性。这种硅掺杂碳材料有潜力成为一种低成本、高效的氧还原反应催化剂,并为设计经济、高性能的非金属催化剂提供了新方法,进一步推动了未来碱性燃料电池设备的发展。
文献:https://doi.org/10.1016/j.carbon.2024.119881
本文来自材料研究前沿,本文观点不代表石墨烯网立场,转载请联系原作者。