
引言
单原子催化剂因其独特的催化活性和极高的原子利用效率,可以有效解决锂硫电池中硫还原反应(SRR)中迟缓的反应动力学问题。作为一种特殊的负载型金属催化剂,单原子催化剂是指活性金属以单个原子形式负载于载体表面。因此,载体对负载的金属单原子的物理化学性质起到关键作用,进而影响催化剂的活性和选择性。由于单原子催化剂主要采用碳材料作为基底材料,SRR反应过程中单原子活性受到了碳原子的单一性和缺乏调节单原子配位环境的限制。此外,金属原子锚定在碳基底材料中,并与氮或碳原子配位,导致金属原子中部分价电子转移到配位环境中,从而影响单原子的催化活性。考虑到硫还原反应是一个涉及多电子的催化反应,构建电子态丰富的单原子活性位点有助于催化SRR。
成果展示
近日,山东师范大学齐思云博士(第一作者)、中国科学院青岛能源所李川川项目副研究员(通讯作者)和山东大学赵明文教授(通讯作者)以理论计算的形式探究了石墨烯/电子化合物异质结负载的单原子催化剂对锂硫电池中硫还原反应的促进作用。电子化合物是一类特殊的以电子作为阴离子的化合物,通过改变单原子与载体之间的电子相互作用,从而改变单原子活性位点对中间体的结合强度。DFT计算表明,电子化合物向石墨烯注入电子,随后转移至单原子金属中。电子将填充到过渡金属原子的d轨道,进而缩短d能带中心与费米能级之间的距离。这种独特的电子结构降低了反键轨道的填充,因此增强了单原子与吸附物之间的键合强度,并且加快硫还原反应的进行。此外,建立的新型描述符可为预测和开发潜在的锂硫催化剂提供理论设计思路。该研究工作以“Single-atom catalysts supported on graphene/electride heterostructures for the enhanced sulfur reduction reaction in lithium-sulfur batteries”为题发表在期刊Journal of Energy Chemistry上。
图文导读
基于电子化合物构建石墨烯/电子化合物异质结
自然界中的物质,其电子通常围绕原子核运动,但有一类特殊的化合物,电子游离于原子核之外,局域在晶格间隙的位置,称为电子化合物。该化合物中的一部分电子不依托特定的元素,独立作为阴离子,使得这些材料往往具有极低的功函数和优异的给电子能力。特别是二维电子化合物(如Y2C和Ca2N),由于其阴离子电子夹在两层阳离子之间,形成受限的二维电子气,引起了广泛关注。因此,我们选择这些具有独特物理化学性质的电子化合物材料作为研究对象,与经典的二维石墨烯材料构建石墨烯/电子化合物异质结。
如图1所示,电子化合物具有较低的功函数,在与石墨烯形成异质结时,倾向于向石墨烯转移电子。根据差分电荷密度图结果,Y2C和Ca2N平均向石墨烯中的每个碳原子转移0.19和0.16个电子。这种巨大的电荷转移导致层间距缩短至2.4 Å,比范德华相互作用要短1 Å,但比典型的共价或离子键长1 Å。电子局域化函数证实了电子化合物外部存在一层松散的近自由层间电子,且石墨烯浸浴在该二维电子云中。平面平均的差分电荷密度图更加突出了电荷的重分布。电子态密度表明电子的转移导致了异质结电子结构费米能级的升高,并在费米能级附近呈现更加丰富的电子态。鉴于电荷转移和准键特性,这种不同于传统“范德华异质结构”的石墨烯/电子化合物异质结被称为“给-受体异质结(donor-acceptor heterostructure)”。
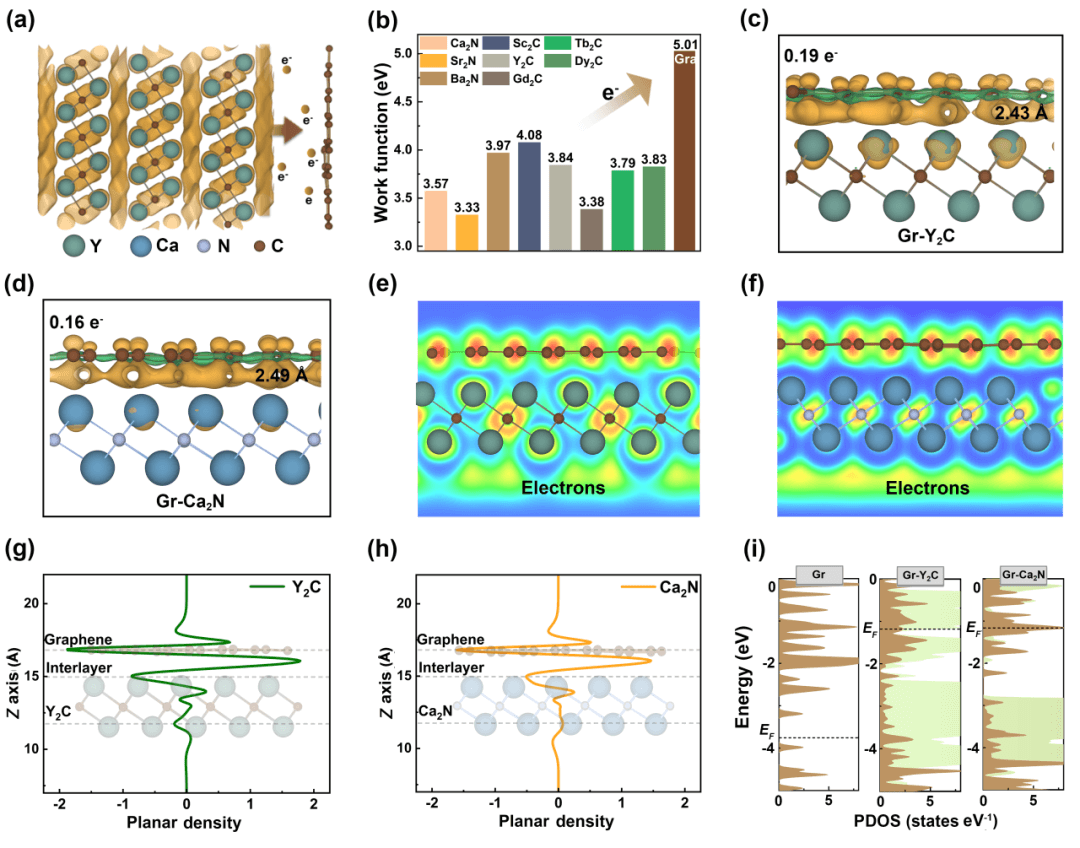

图 1. 石墨烯/电子化合物(Gr-electride)异质结构图。(a)电子化合物向石墨烯注入电子图示。(b)不同电子化合物的功函数。(c-f)Gr-Y2C和Gr-Ca2N的差分电荷密度图和电子局域密度图。(g-h)Gr-Y2C和Gr-Ca2N在z方向上的电荷平面平均密度图。(f)电子态密度图。
筛选单原子催化剂
作者首先选取了9种3d过渡金属单原子(Ti, V, Cr, Mn, Fe, Co, Ni, Cu)进行研究(图2),通过计算形成能表明金属原子在异质结上存在较强的热力学稳定性。通过电子态密度和Bader电荷分析可知,异质结中的金属单原子上存在更多的电子,并具有更高的d能带中心,这是由电子化合物表面的近自由电子通过石墨烯转移至过渡金属单原子所导致的。通过吸附能的计算,确定了异质结可改善单原子对硫物种的吸附能力,其中Sc、Ti、V单原子改善效果最为显著,因此是最有潜力的SRR催化剂。
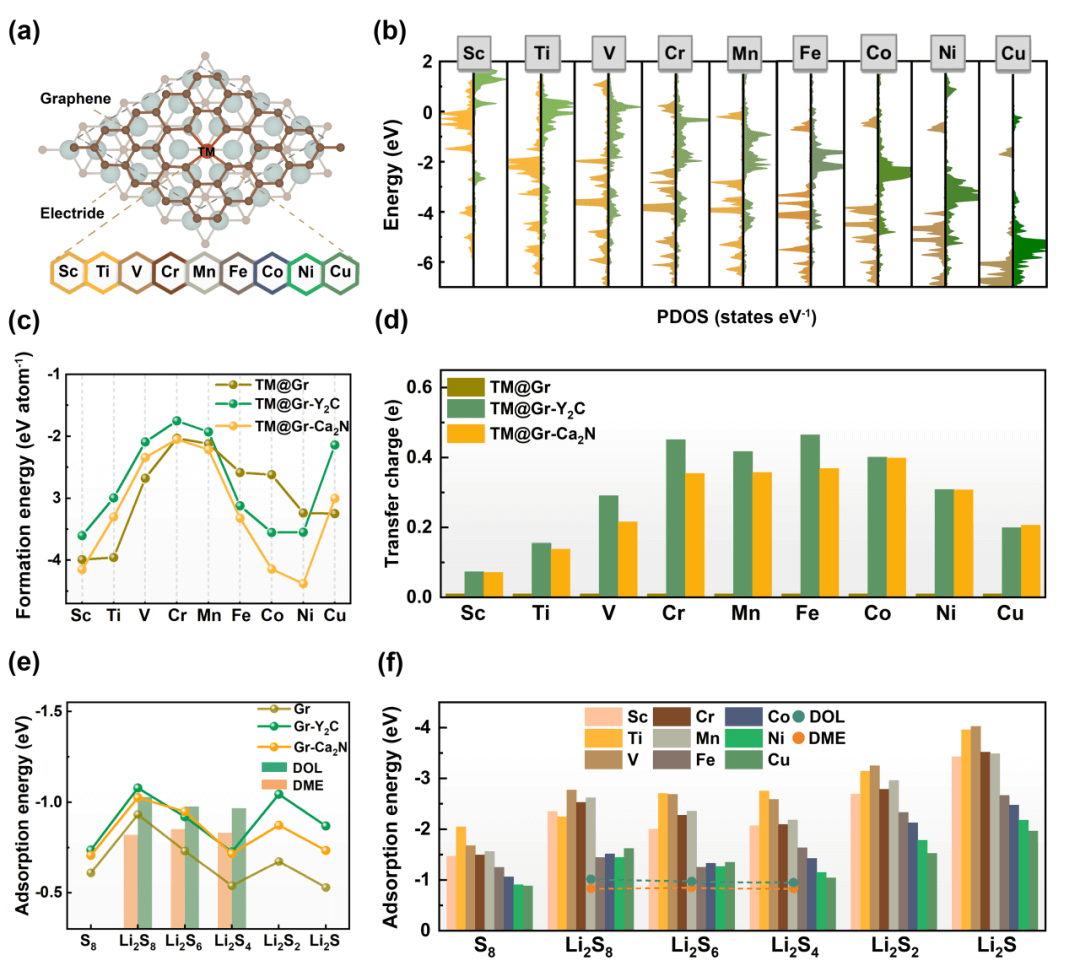
图 2. 过渡金属原子负载的石墨烯/电子化合物异质结(TM@Gr-Y2C)的稳定性和锂硫分子吸附行为。(a)TM@Gr-Y2C的晶格结构。(b)未添加电子化合物(左栏)和加入电子化合物(右栏)体系的过渡金属原子的电子态密度图。三类体系中过渡金属原子的(c)形成能和(d)金属原子表面的电荷量。(e)锂硫分子的吸附能对比。(f)TM@Gr-Y2C中吸附锂硫分子的吸附能。
吸附键强分析和电荷转移
如图3所示,作者选取吸附能力最强的V@Gr-Y2C结构,以解释电子化合物加入提高吸附能力的原因。根据分子轨道理论和d带中心分析,在加入电子化合物后,过渡金属原子中d能带中心的位置上移,从而减低与费米能级之间的能量差。这种变化导致了反键轨道的升高,使更多的反键轨道高于费米能级,减少了反键轨道的填充,进而增强了单原子与锂硫分子的吸附强度。晶体轨道哈密顿布局(COHP)提供了原子间化学键强度和性质信息,积分COHP(ICOHP)作为测量化学键强度的定量方法,其绝对值越大表示原子间键强度越强。根据绘制出的ICOHP图,V@Gr-Y2C (−2.95 eV) 和V@Gr-Ca2N (−2.91 eV) 与S的键合强度明显高于V@Gr (−2.34 eV),这是其吸附性能增强的关键原因。Bader电荷和键长分析说明引入电子化合物改变了过渡金属活性位点处的电子状态,不仅促进了吸附的锂硫分子的电荷转移,而且起到活化S8分子反应物的作用。
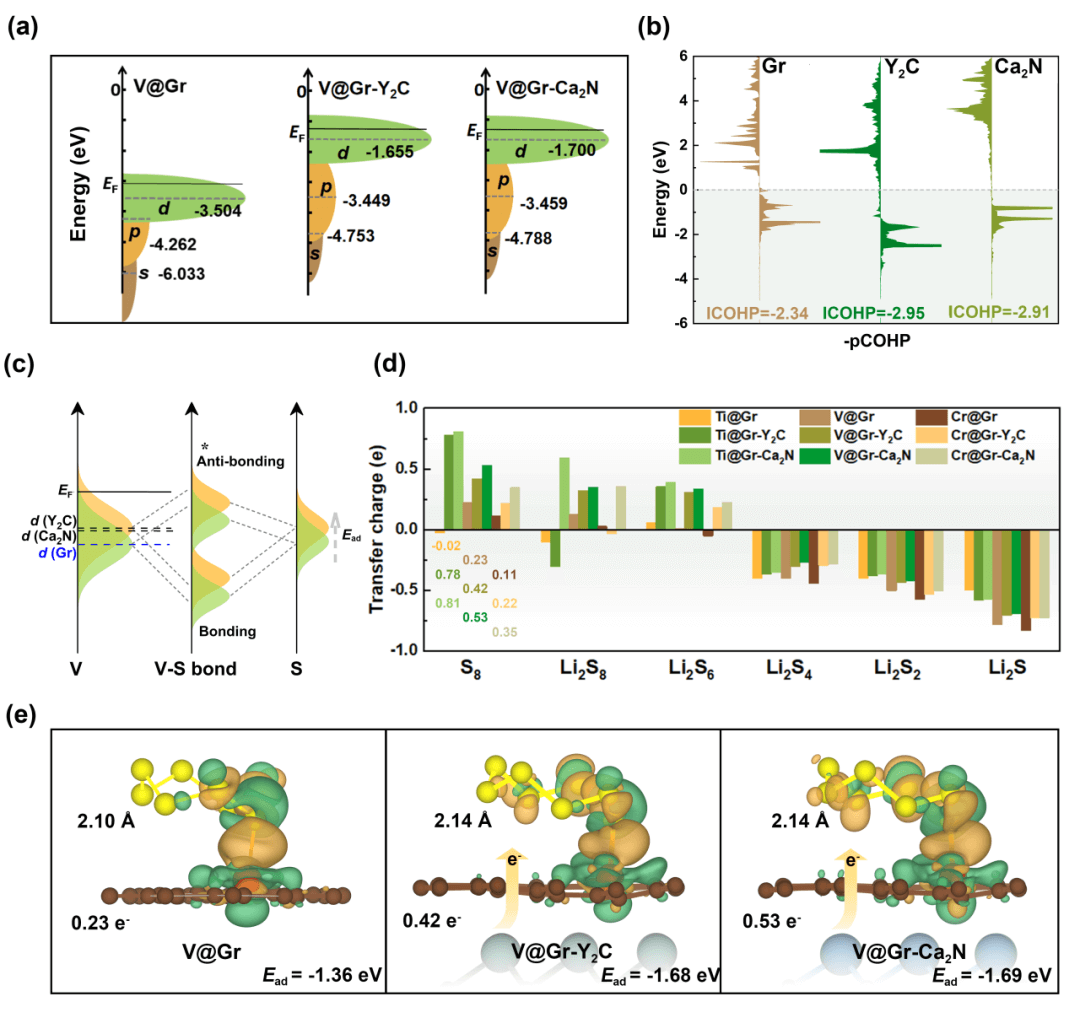
图3 TM@Gr-electride中的锂硫分子吸附行为。(a)V原子负载的不同体系中的s、p、d能带中心的能级示意图。(b)V-S键的COHP。(c)锂硫分子吸附物与V@Gr,V@Gr-Y2C和V@Gr-Ca2N的相互作用示意图。(d)Bader电荷分析。(e)S8分子在V@Gr,V@Gr-Y2C和V@Gr-Ca2N的差分电荷密度图。
硫还原自由能分析和Li2S解离
进一步系统地讨论Ti、V和Cr单原子体系中硫还原反应路径(S8* → Li2S8* →Li2S6* → Li2S4* → Li2S2* → Li2S*)中吸附物种自由能之间的关系,发现前三步反应是放热过程,而后两步反应则为吸热过程,其中最后一步(Li2S2* → Li2S*)为速控步。因此ΔG (Li2S*)可被视为描述SRR活性的重要能量参数。从图4中可以很明显的看出,相比石墨烯负载的单原子催化剂,引入电子化合物后的单原子催化剂能显著降低速控步的吉布斯自由能垒,提高SRR的性能。此外,随着电子化合物的引入,Li2S的解离能垒也随之降低,从而表现出更快的反应速率。
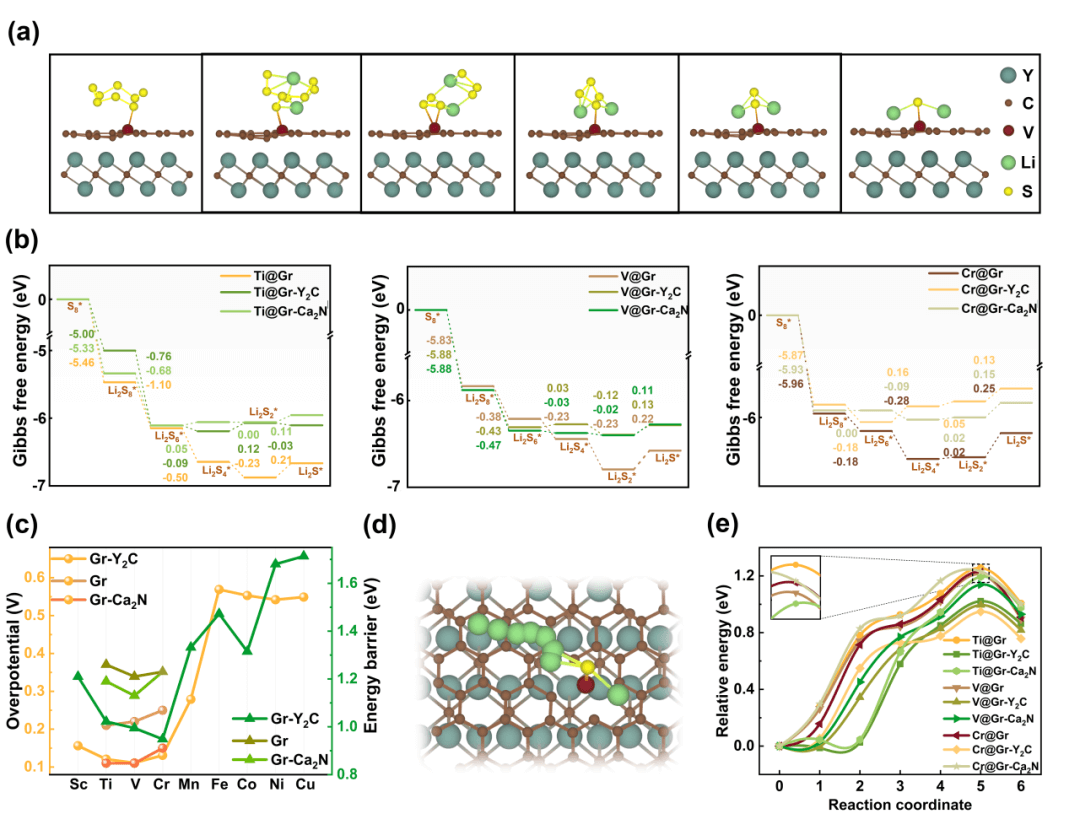
图4. 硫还原反应和Li2S的解离过程。(a)锂硫分子的反应吸附结构图。(b)硫还原反应的吉布斯自由能图。(c)反映过电势(左栏)和Li2S分解势垒(右栏)示意图。Li2S分解的(d)反应路径和(e)能量势垒。
构建描述符识别催化活性来源
最后,作者选取了通用的描述符以评估其催化性能,并采用Pearson相关系数和显著性水平系数构建了热力图,用于评估d能带中心、电荷转移、功函数、ICOHP、键长、范德华力作用占比等可能的因素与过电势、吸附能、Li2S解离势垒等决定催化性能的因素之间的关系(图5)。结果表明,S8分子吸附的范德华力作用占比与催化性能之间存在最强相关性,且过渡金属与硫原子的成键强度(ICOHP)与催化性能密切相关。S8在吸附过程中的范德华力作用占比直接反映了基底材料捕获S8分子的能力,而电子化合物对过渡金属的给电子效应促进了活性位点对S8分子的吸附,这类独特的描述符为评价锂硫电池的催化性能奠定了坚实的基础。
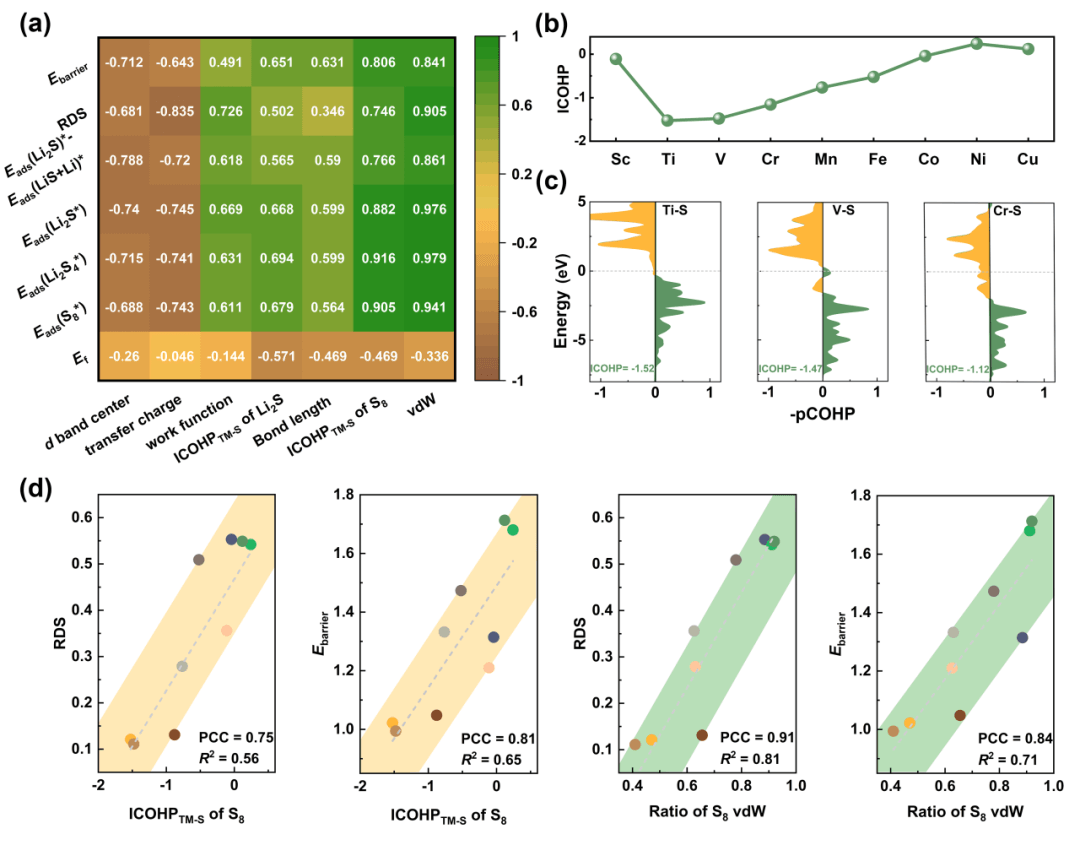
图 5. 描述符的构建。(a)潜在因素与决定催化性能之间的相关系数矩阵热图。(b)TM-S键的ICOHP数值图。(c)S8吸附在Ti@Gr-Y2C、V@Gr-Y2C和Cr@Gr-Y2C中TM-S键的pCOHP和ICOHP示意图。(d)线性相关关系。
小结
该工作构建了石墨烯/电子化合物异质结作为单原子催化剂的载体,用于调节单原子的SRR催化活性。电子化合物表面的电子云转移至石墨烯,然后传递到金属单原子,影响金属活性位点的电子结构和与锂硫分子的结合强度。通过计算吸附能、吉布斯自由能、过电势和Li2S分解势垒,进一步揭示了电子化合物对单原子催化锂硫反应的促进作用。同时构建相应的描述符用以阐明高催化活性的来源。这一研究为锂硫电池电催化剂的理论设计提供了新思路。
文章信息
Single-atom catalysts supported on graphene/electride heterostructures for the enhanced sulfur reduction reaction in lithium-sulfur batteries
Siyun Qi, Chuanchuan Li*, Gang Chen, Mingwen Zhao*
J. Energy Chem.
DOI: 10.1016/j.jechem.2024.07.013
作者信息
齐思云,山东师范大学物理与电子科学学院讲师。从事纳米材料模拟第一性原理计算的理论研究,涉及电子输运、光电催化及能源转换以及理论模型构建等。在Sci. Bull.,Phys. Rev. B,J. Phys. Chem. Lett.和J. Energy Chem.等期刊发表第一作者或通讯作者论文多篇,发表SCI收录论文20余篇。
李川川,中国科学院青岛生物能源与过程研究所项目副研究员。主要从事高比能锂硫电池和低成本钠钾电池的研究,包括电极材料和电解液及其界面等。以第一/通讯作者在能源、材料和化学等方面的国际权威杂志如Adv. Energy Mater.、Nano Today、Energy Storage Mater.、Adv. Funct. Mater.和J. Energy Chem.等发表文章SCI论文20余篇,被他人引用1000余次。
赵明文,山东大学物理学院教授,副院长。长期从事凝聚态理论和材料计算研究,在低维材料的电子结构和缺陷效应等方面取得了多项研究突破,解决了低维材料器件应用中的一些基础问题,提出的理论模型被国内外多个实验课题组验证,并成功地实现了器件应用,先后承担完成了国家973、国家自然科学基金重大研究计划、重点项目和面上项目等课题,形成了且具有一定特色和影响力研究方向。累计在Phys. Rev. Lett.,Adv. Mater. 和Nano Lett. 等著名学术期刊上发表SCI 论文 360余篇,被SCI引用10000余次,2014−2024年连续多年入选“中国高被引学者(物理天文类)”。
本文来自JEnergyChem,本文观点不代表石墨烯网立场,转载请联系原作者。