
研究背景
过渡金属单原子催化剂(SACs)具有高催化活性、选择性和稳定性,这使它们成为燃料电池、水裂解和其他能源转换技术中的有前景的候选材料。然而,在实际应用中,SACs的性能高度依赖于其结构和化学环境,尤其是碳载体上的异原子配位情况。传统方法中,SACs的制备通常依赖于高温热解过程,其中碳载体与过渡金属前体和异原子源共热解。然而,这种方法存在着一个普遍的问题,即在高温下碳载体易于结构崩塌,导致异原子的排列难以精确控制,从而影响了催化性能的优化。
研究内容
因此,为了克服这一挑战,法国南特大学Romain Gautier教授、扬州大学化学与化学工程学院蒋腾飞和Jingqi Tian教授联合引入了一种拓扑异原子转移策略,旨在在热解过程中防止碳载体结构的崩塌,并精确控制碳载体上的异原子配位。通过利用石墨烯量子点(GQDs)作为碳载体的前体,并在其中自组装出具有腔的纳米结构,科学家们成功地将异原子(例如氮和氧)与金属离子形成配位络合物,从而实现了对异原子排列的精确控制。通过这种方法,科学家们成功地制备出具有优异电催化性能的过渡金属单原子催化剂,为电化学和催化领域的应用提供了新的可能性。以上成果在Nature Communications发题为“Tailoring coordination environments of single-atom electrocatalysts for hydrogen evolution by topological heteroatom transfer”研究论文。
图文导读
为了研究氨基和羟基功能化的石墨烯量子点(NGQDs)在碱性介质中的自组装行为以及相关结构特征,研究者进行了图1的实验。图1包括了螺旋纤维的结构表征结果。在低放大倍数的扫描电子显微镜(SEM)图像中(图1a),可以看到螺旋纤维的整体形态。高放大倍数的SEM图像(图1b)显示了纤维表面的细节结构。透射电子显微镜(TEM)图像(图1c)进一步展示了纤维的形态。高分辨透射电子显微镜(HRTEM)图像(图1d)揭示了纤维的晶体结构。原子力显微镜(AFM)图像(图1f)展示了纤维的高度分布。傅立叶变换红外光谱(FT-IR)谱图(图1g)展示了NGQDs和螺旋纤维之间的化学结构差异。时间依赖的光致发光(PL)光谱(图1h)描述了NGQDs在自组装过程中的光学特性变化。
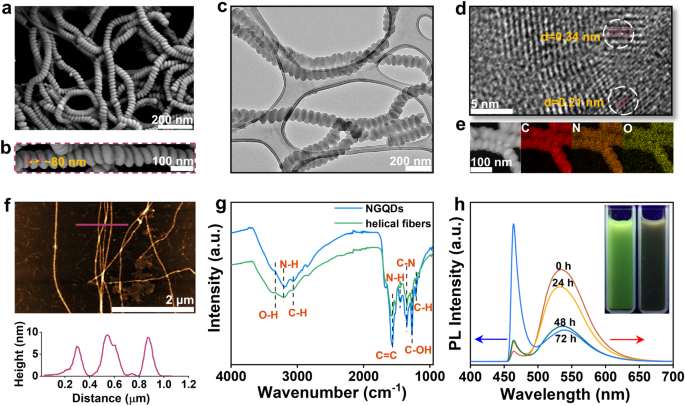

图 1 | 螺旋纤维的结构表征。
图2显示了不同pH条件下NGQDs的自组装行为和产物形貌。图2a通过示意图形象地展示了在不同pH条件下NGQDs的自组装行为,以及形成的纳米片或螺旋纤维结构。分子动力学(MD)模拟结果(图2b)提供了NGQDs在不同pH条件下自组装过程中的能量变化信息。ζ电位测量结果(图2c)反映了不同pH条件下NGQDs自组装体的表面电荷状态。此外,图2还展示了在不同pH条件下的荧光光学图像,显示了组装体的形貌和结构特征。这些研究结果对于理解碱性条件下NGQDs的自组装机理以及pH对组装产物形态的影响具有重要意义。
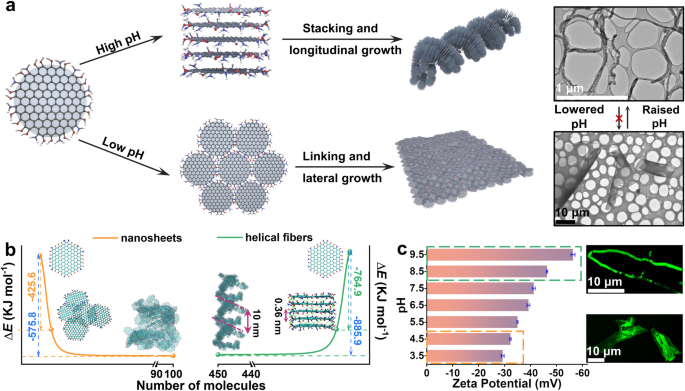
图 2 | 基于GQD的组装机理研究。
图3展示了研究者设计的合成和表征Co-PxN4-x-C单原子催化剂(SACs)的过程。首先,他们通过组装-磷化方法成功合成了具有磷和氮共配位的碳纤维结构(Co-PN-C)。在这一过程中,功能团和金属离子之间发生了螯合反应,将氮、氧与Co2+螯合在一起,形成了前驱体。接着,研究者通过磷化反应将前驱体转化为具有P和N共配位的碳纤维,即Co-PN-C。通过对不同温度下的热处理,发现500°C至700°C范围内,螺旋纤维形貌可以得以保留,但在800°C时会崩溃形成碎片颗粒。进一步的分析显示,经过磷化处理后的样品在TEM和HAADF-STEM图像中呈现出均匀分布的Co、P、N和C元素,表明Co原子被均匀分散在碳纤维中。XPS和XANES分析进一步证实了Co原子的稳定价态和化学环境,而EXAFS结果显示了Co原子与P和N原子的配位情况。这些结果表明,通过组装-磷化方法可以有效地合成具有P和N共配位的Co单原子催化剂。
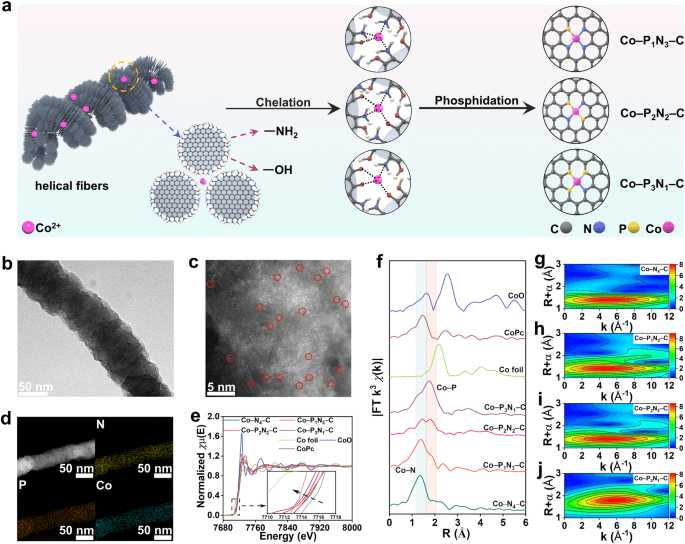
图 3 | Co–PxN4-x–C SACs的合成和表征。
图4展示了研究者对Co-PxN4-x-C(x=0-3)SACs的电催化性能进行的评价。在1.0 M KOH电解液中进行的电化学测试显示,Co-PxN4-x-C催化剂的氢析出反应(HER)性能表现出一定的规律性。具体而言,Co-P2N2-C的性能最佳,其过电位最小,达到100 mA/cm2的电流密度时,过电位(η100)为131 mV,且具有最小的Tafel斜率。电化学阻抗谱(EIS)结果显示,Co-P2N2-C具有较快的HER动力学过程。此外,TOF研究结果表明,在200 mV的过电位下,Co-P2N2-C的TOF值最高,进一步验证了其出色的HER活性。最后,研究者利用Co-P2N2-C催化剂组装了一个基于阴极交换膜的水电解装置,并评估了其在水电解过程中的稳定性和效率。结果显示,该装置在1.0 A/cm2的电流密度下工作100小时,并且具有较高的氢气产率和法拉第效率。
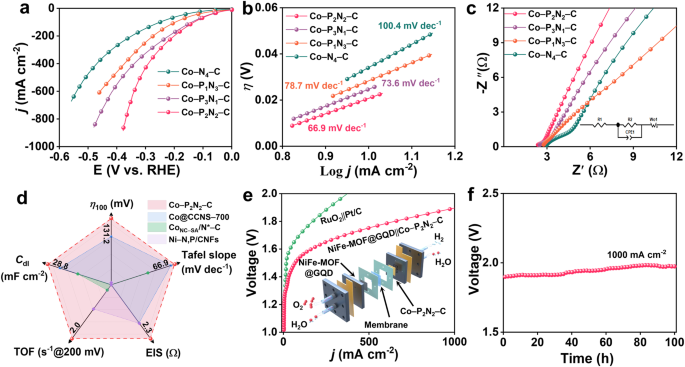
图 4 | 在1.0M KOH电解液(pH=14)中HER性能的电化学表征。
图5a展示了电子密度差异的顶视图和Co-N4-C、Co-P1N3-C、Co-P2N2-C以及Co-P3N1-C的Bader电荷图。结果显示,在Co-PxN4-x-C中,钴原子周围的电子富集状态相比于Co-N4-C更为明显,钴原子的电荷状态也更为降低,这表明P的配位有助于降低钴中心的氧化态。此外,图5b至图5e展示了水解过程自由能、氢吸附能以及相关能量计算结果,表明Co-P2N2-C具有最优的催化性能。图5f和图5g展示了从头开始的分子动力学模拟结果,揭示了催化剂的结构耐久性和Co-N、Co-P键长度的统计。这些结果有助于理解磷原子在Co-PxN4-x-C催化剂中的作用机制,为设计更高效的催化剂提供了重要参考。
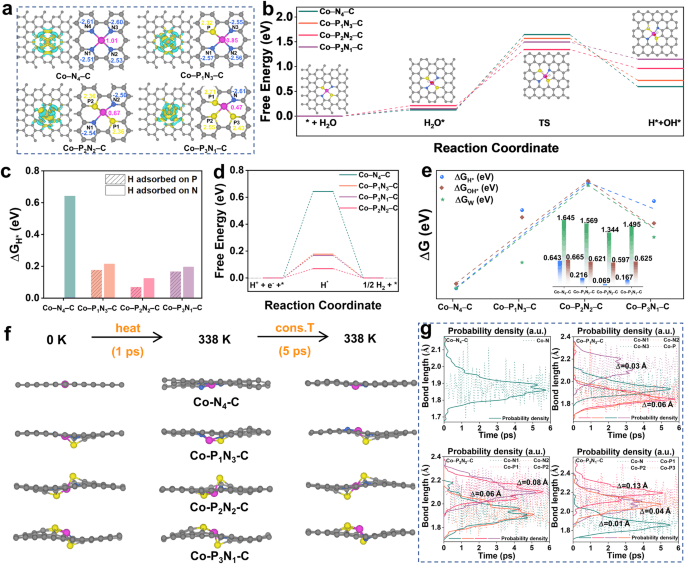
图 5 | 理论研究。
结论展望
本文提出了一种新颖的异原子转移策略,通过自组装方法合成并系统调控磷配位的钴单原子催化剂(Co–PxN4-x–C)。这一策略利用了石墨量子点的自组装特性,使得催化剂的结构可以被保留,并且可以将螯合环境中的杂原子定向转移成单原子催化剂。通过光谱分析和理论计算,研究者发现引入特定配位的磷原子能够优化催化剂表面的电子密度,从而提高氢析出反应的活性。
这项工作为理解和调控单原子催化剂的结构-活性关系提供了新的思路和方法。此外,本研究强调了精确控制配位环境对催化性能的影响,为未来设计高效电催化剂提供了重要的指导和启示。这种策略的成功应用展示了在碳超结构中精确调控配位环境的巨大潜力,为开发更高性能的能源转换和存储技术奠定了基础。
该工作发表在Nature Communications上
文章链接:https://doi.org/10.1038/s41467-024-47061-6
本文来自低维材料前沿,本文观点不代表石墨烯网立场,转载请联系原作者。