
2024年1月31日,J. Am. Chem. Soc.在线发表了克罗地亚萨格勒布大学Igor Rončević课题组的研究论文,题目为《Electronic Structure of Metalloporphenes and Antiaromatic Analogues of Graphene》。
锌卟啉烯(zinc porphene)是一种由四方晶格中完全融合的锌卟啉构成的二维材料。它有一个完全共轭的π体系,使其类似于石墨烯。锌卟啉烯最近被合成,粗略的电导率测量以及红外和拉曼光谱的结合都表明它是一种半导体。这与之前对其电子结构的所有预测形成了对比,这些预测表明了金属导电性。
在此研究中,作者证明锌卟啉烯的能隙打开是由其晶胞从正方形到矩形的Peierls畸变引起的,从而首次给出了与实验一致的电子结构。考虑到这种畸变需要对电子离域进行适当的处理,这可以使用具有大量精确交换的混合泛函来完成。然后,将这样的泛函PBE38应用于预测许多第一过渡行金属卟啉烯的性质,其中一些已经制备好。
研究发现改变金属会强烈影响金属卟啉烯的电子结构,从而产生丰富多样的金属导体和半导体,这可能会引起分子电子学和自旋电子学的极大兴趣。这些材料的性质主要由Peierls畸变的程度和π体系中的电子数决定,类似于在氧化或还原时在环状共轭分子中观察到的芳香性变化。这些结果说明了如何将反芳构性的概念推广到周期体系。
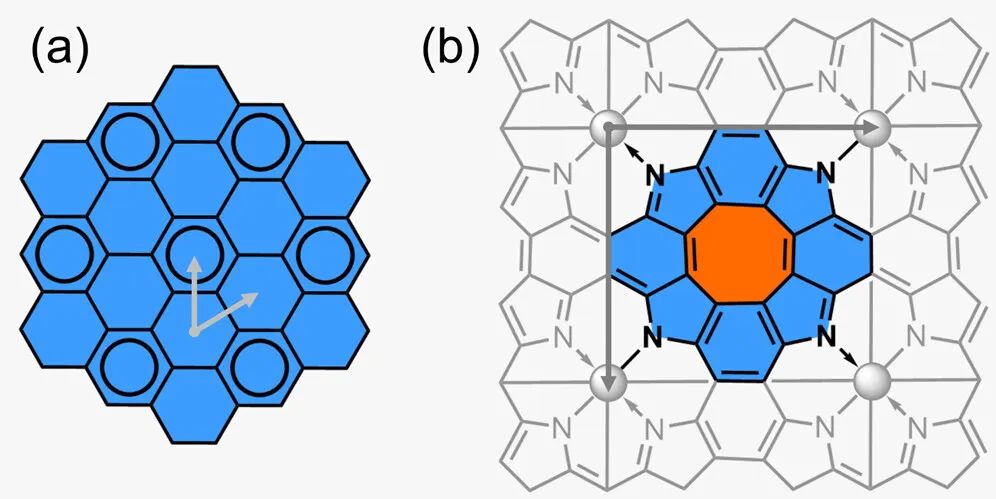

图1 石墨烯和金属卟啉烯(MP)的结构
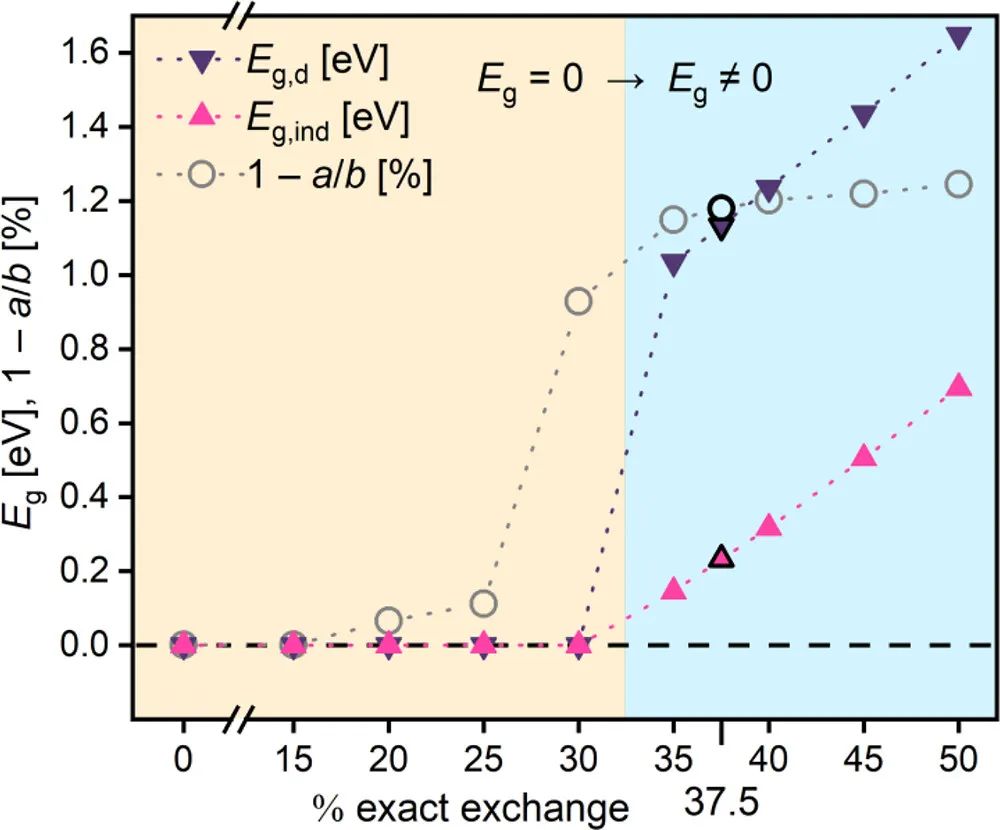
图2 ZnP在不同交换关联(EE)水平下的直接带隙和间接带隙及晶胞不对称性
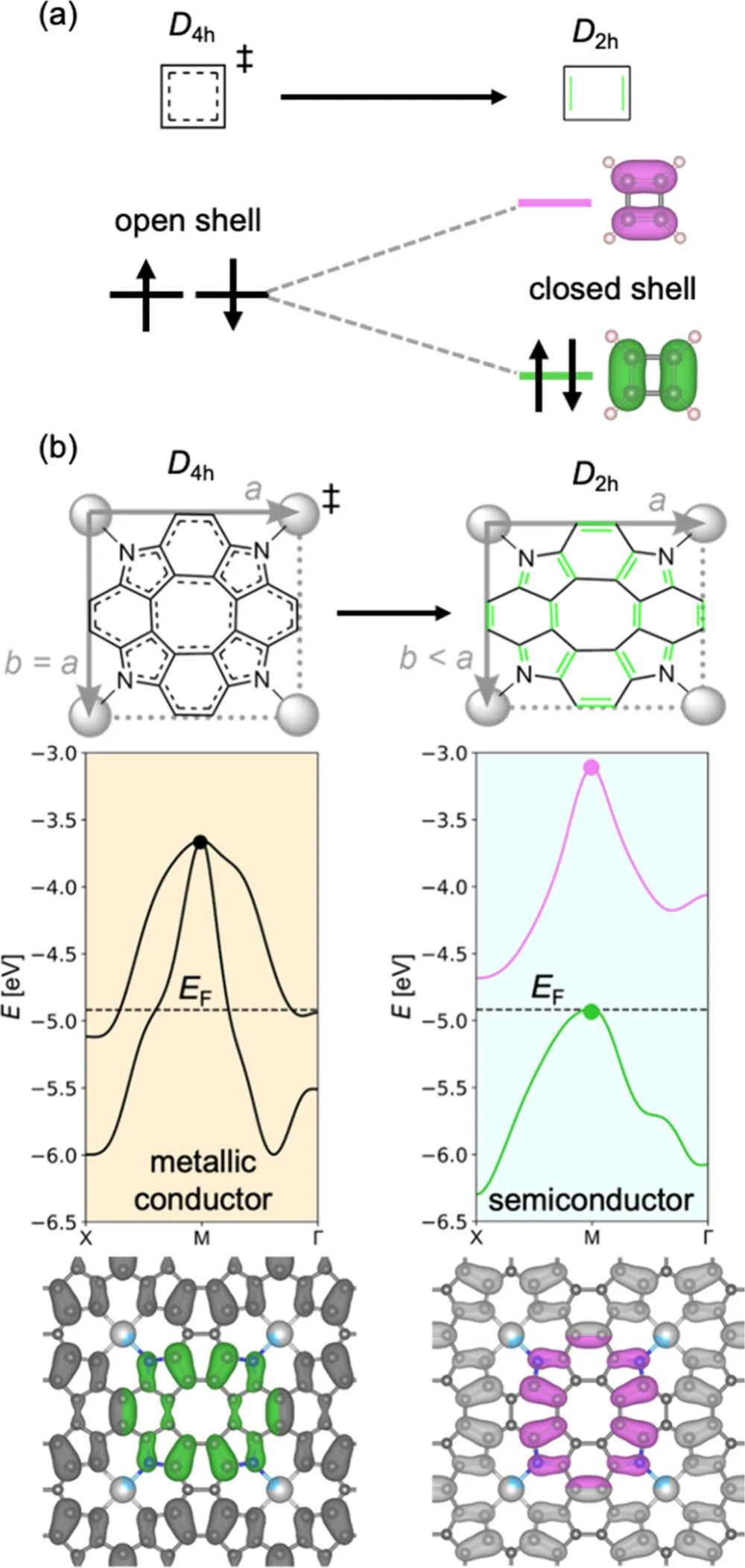
图3 环丁二烯的对称性破缺和ZnP中的Peierls畸变
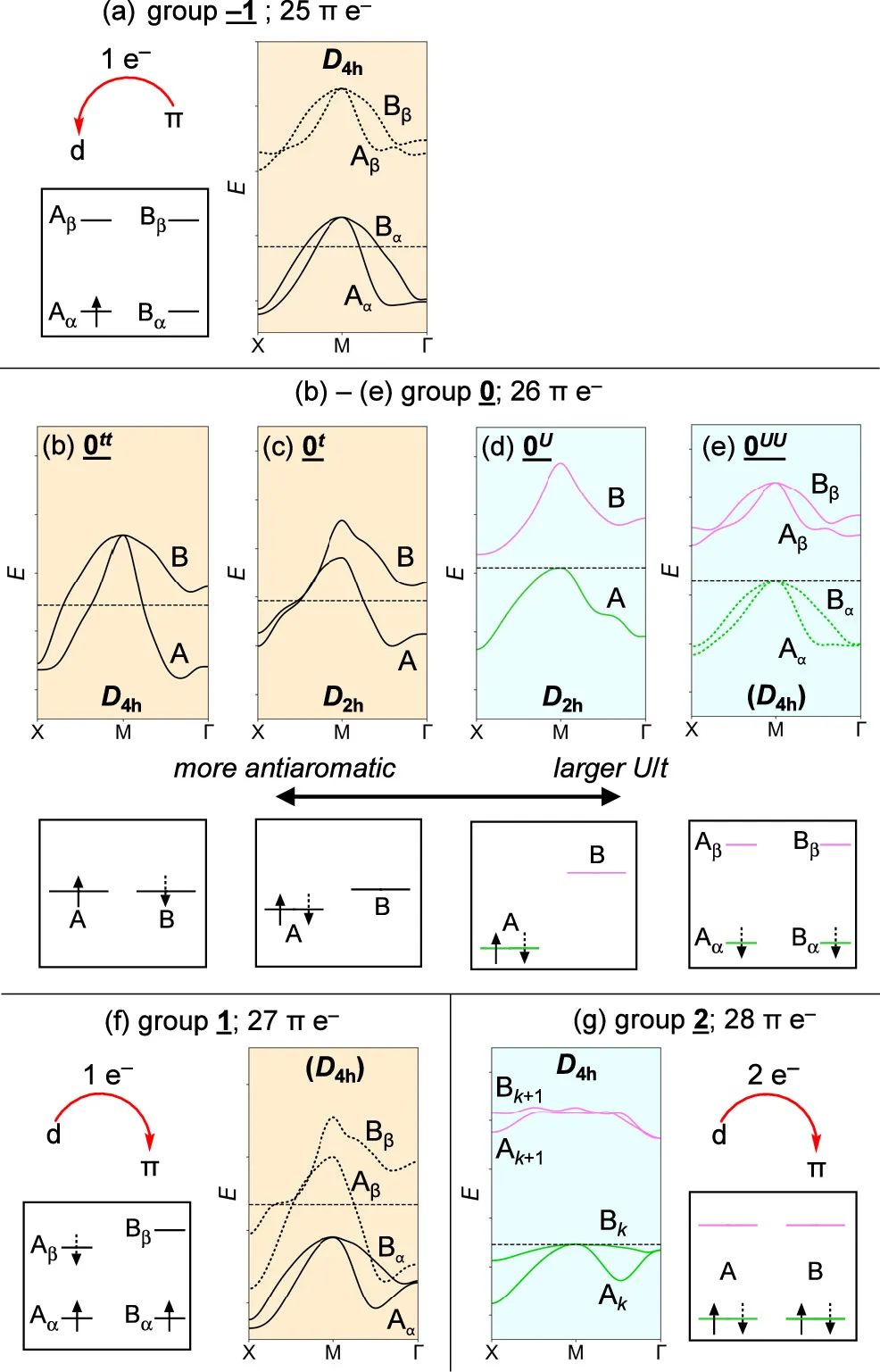
图4 金属卟啉烯的最小能带结构和类似MO图
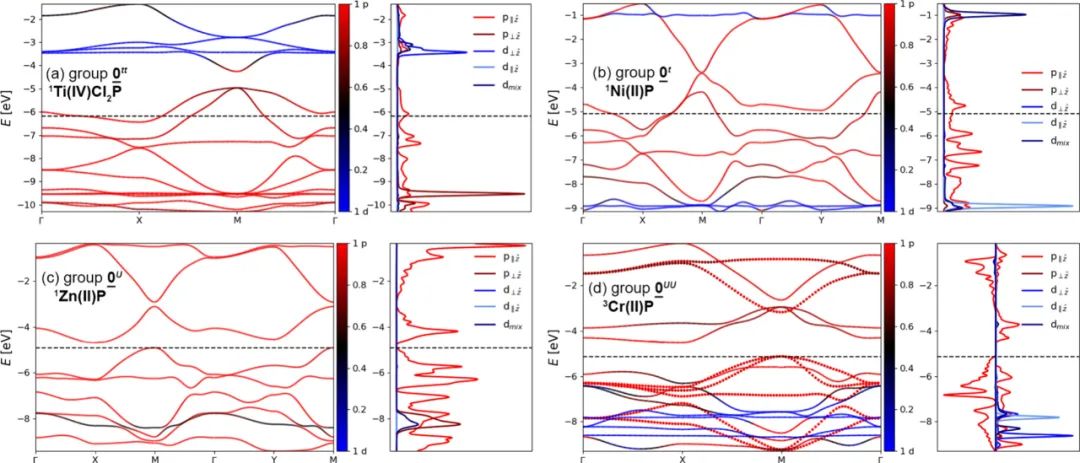
图5 能带和态密度图
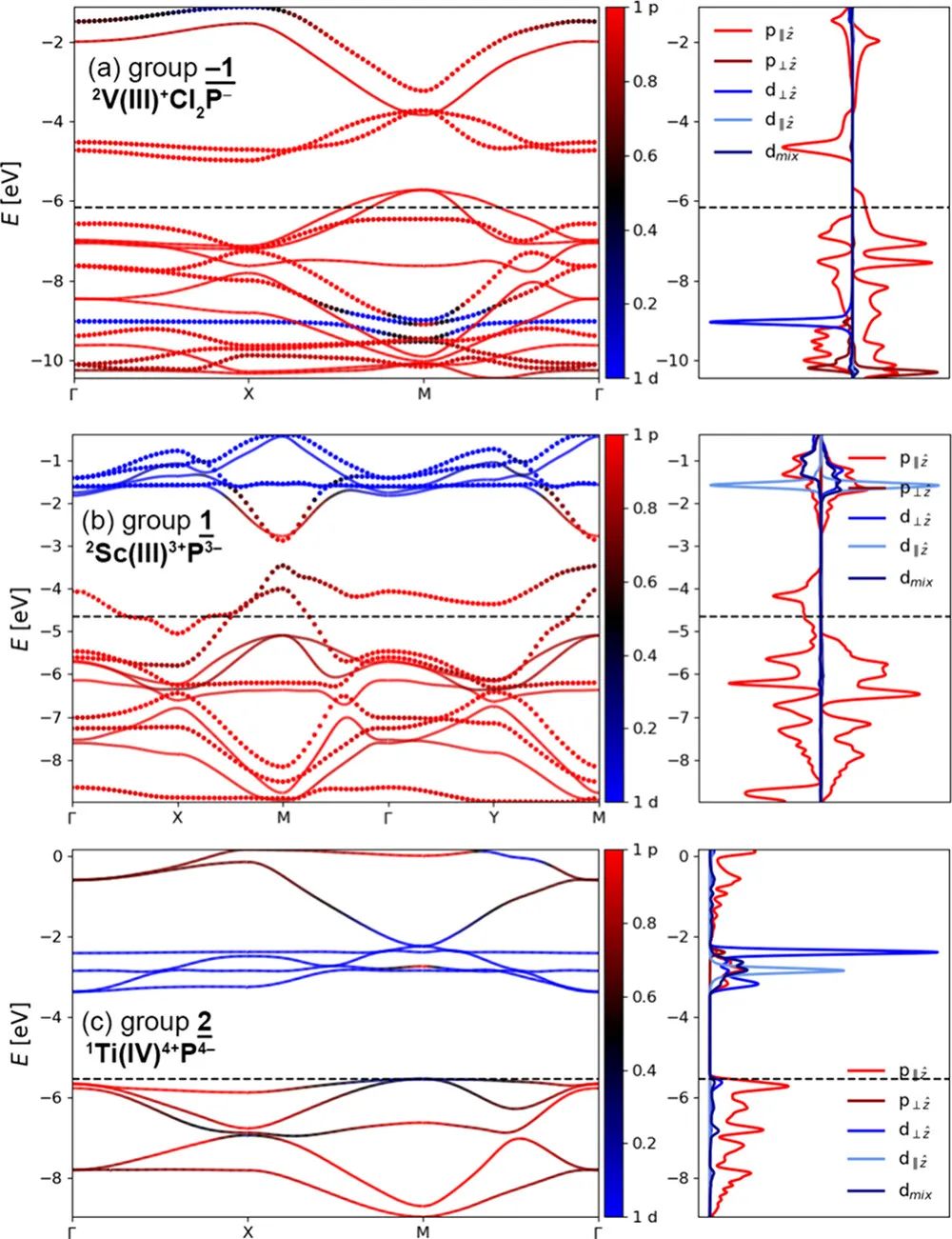
图6 能带和态密度图
【论文链接】
Pavlak, I., Matasović, L., Buchanan, E.A. et al. Electronic Structure of Metalloporphenes and Antiaromatic Analogues of Graphene. J. Am. Chem. Soc., 2024. https://doi.org/10.1021/jacs.3c12079
【其他相关文献】
[1] Matochová, D., Medved’, M., Bakandritsos, A. et al. 2D chemistry: Chemical control of graphene derivatization. J. Phys. Chem. Lett., 2018, 9, 3580–3585. https://doi.org/10.1021/acs.jpclett.8b01596
[2] Magnera, T.F., Dron, P.I., Bozzone, J.P. et al. Porphene and porphite as porphyrin analogs of graphene and graphite. Nat. Commun., 2023, 14, 6308. https://doi.org/10.1038/s41467-023-41461-w
本文来自科研任我行,本文观点不代表石墨烯网立场,转载请联系原作者。