
二维(2D)材料石墨烯及其衍生的纳米结构的合成开辟了化学、物理和材料科学交叉的跨学科领域。在这个领域,来自分子或扩展固态系统的直觉是否决定了这些材料的物理性质,这是一个悬而未决的问题。在这项工作中,本研究使用从头算模拟技术研究了电场中二维扶手椅石墨烯纳米带上原子的电迁移力。研究结果表明,力与吸附质-表面键中的诱导电荷有关,而不仅仅与诱导的原子电荷有关,并且左右有效键序可以用来预测力的方向。特别关注3d过渡金属原子,本研究展示了苯上金属原子的简单模型如何解释无机化学图片中的力。这项研究表明,电场中二维表面上的原子迁移是由一幅不同于电场中带电粒子常用静电描述的图片决定的,因为潜在的键合和分子轨道结构与电迁移力的定义相关。因此,包括原子配体场的扩展模型可以更好地理解非平衡条件下吸附质在表面上的扩散。
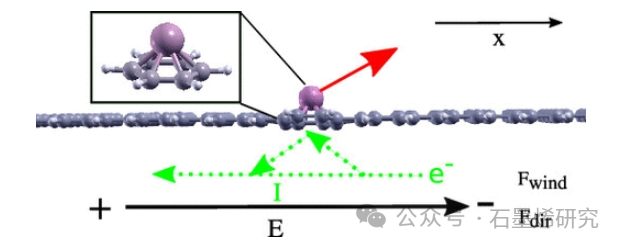

图1. 在沿表面方向(“横向”)和电流I施加的电场E中,位于2D碳纳米结构顶部的单个原子。原子经历电迁移力(红色箭头),该电迁移力由电场引起的贡献Fdir和电流中电子散射引起的贡献F wind组成。插图显示了吸附原子和苯环,代表了扩展结构的简化模型。
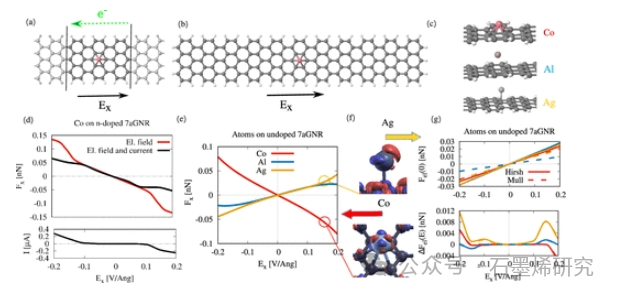
图2. 在横向电场中/在经历电迁移力的有限偏压下吸附在7-aGNR上的单原子。(a) 具有半无限大aGNR电极的7-aGNR上的Co原子。在左电极和右电极之间施加偏置电压,从而产生电场和电流。(b) 横向电场Ex.中有限7-aGNR上的Co原子。(c)单原子(Co、Al和Ag)在7-aGNR:Co和Al更喜欢中空位置,而Ag更喜欢顶部位置。(d) 顶部:来自(a)所示传输设置的Co原子上的电流和场诱导力Fx(黑线),以及来自(b)所示设置的纯场诱导力(红线)。底部:(a)在电场上流过系统的电流。(e) 电场中7-aGNR上Co、Al和Ag原子上的力。(f) 当Ex=0.15V/Å时,Ag和Co上的场诱导电荷密度。(g) 顶部:通过Hirshfeld(实线)和Mulliken(虚线)分析获得的,在GNR上吸附时,由于Co、Al和Ag上感应的净电荷Q(0)而产生的静电力Fel(0)。底部:电场Ex上的场感应电荷产生的静电力ΔFel(E)ΔQ(E)。
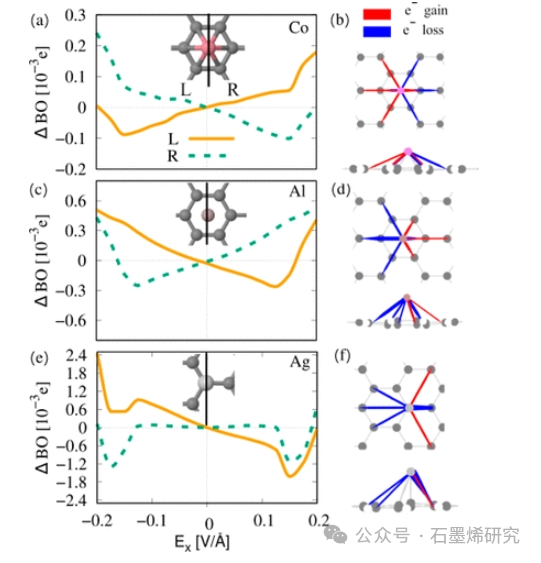
图3. 分析横向电场Ex.中有限7-aGNR上Co、Al和Ag的键序变化(ΔBO)以及左右键中的电荷再分配。(a)电场Ex.上7-aGNR上Co的左右场相关ΔBO。插图显示了左右键的划分。(b) Co在Ex=0.15eV/Å时诱导键电荷的可视化(俯视图和侧视图),其中蓝色表示电子耗尽和红色积聚。(c,d)对中空位点上的Al进行相同分析,对顶部位点上的Ag进行相同分析。ΔBO描述的原子和表面之间键中的电荷再分配预测了场诱导力在原子上的方向。
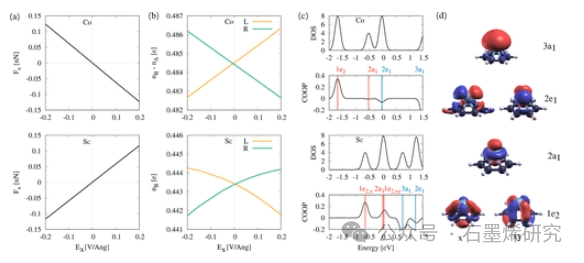
图4. 电场Ex.中Co和Sc在苯上的电迁移力、成键和反键电子以及能态的比较(“C6H6+M”模型)。(b) 场诱导的成键和反成键电子,nB和nA,在吸附原子的左侧和右侧。而对于C6H6+Sc,只有键合轨道被占据(nA=0),C6H6+Co表现出来自反键轨道2e1的电子密度。(c) 对于Co(顶部)和Sc(底部),C6H6+M的态密度(DOS)和吸附原子和C6H6之间的晶体轨道重叠布居(COOP)。在COOP中,显示了C6H6+M态的能量位置。COOP表示态的性质(正态=成键,负态=反键)。(d) C6H6+M分子轨道,根据它们的对称性来表示。
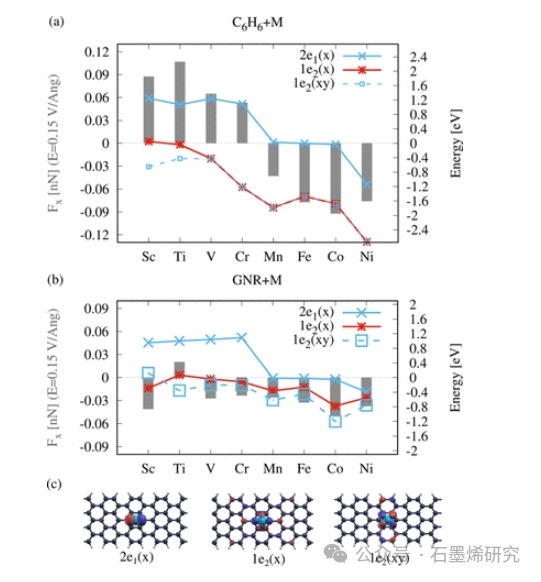
图5. 电迁移力和能态之间的相关性接近EF=0eV。(a)场Ex=0.15V/Å时吸附原子上的力Fx(灰色条),以及苯上原子(C6H6+M)和7-aGNR上原子(GNR+M)的2e1(x)(蓝色)、1e2(x)和1e2(xy)(蓝色虚线)的能态位置。(c) 7-aGNR上Co的分子轨道2e1(x)、1e2(x)和1e2(xy)。早期TM上的力与1e2态的种群有关,而晚期TM由2e1(x)态主导,C6H6+M和GNR+M都更接近EF。
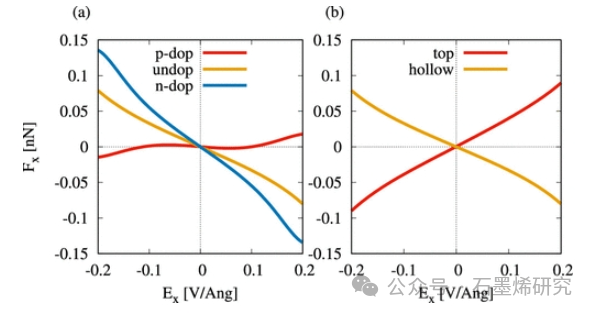
图6.横向电场Ex中7-aGNR上的Co原子上的电迁移力,取决于掺杂水平和原子位置。(a) 在n-、p-和未掺杂的7-aGNR上的Co原子上的电场Ex上的力Fx。(b) 未掺杂7-aGNR的顶部和中空部位上的Co上的力Fx与电场Ex的比较。
相关研究成果由哥本哈根大学Gemma C. Solomon等人2023年发表在JACS Au (链接:https://doi.org/10.1021/jacsau.3c00622)上。原文:Electromigration Forces on Atoms on Graphene Nanoribbons: The Role of Adsorbate–Surface Bonding
本文来自石墨烯研究,本文观点不代表石墨烯网立场,转载请联系原作者。