
众所周知,双金属原子的活性与其化学配位环境密切相关。河海大学姜全国、北京师范大学敖志敏等人通过DFT计算全面研究了含三碳空位和四碳空位的缺陷石墨烯负载Cu双原子催化剂上CO的氧化反应。
计算方法
作者采用DMol3模块进行密度泛函理论(DFT)模拟,并采用含有PBE泛函的GGA来描述交换关联作用,以及采用Grimme方案来描述范德华力。作者将能量、最大力和位移的收敛标准分别设置为10−5 Hartree、0.002 Hartree/Å和0.005Å,并采用了LST/QST和NEB方法来寻找CO氧化反应的最小能量途径。作者使用了具有三维周期边界的4×4石墨烯超胞模型,并将k点设置为5×5×1。作者使用CASTEP代码来进行电子轨道计算,并采用了GGA-PBE泛函和340eV的截断能。
结果与讨论
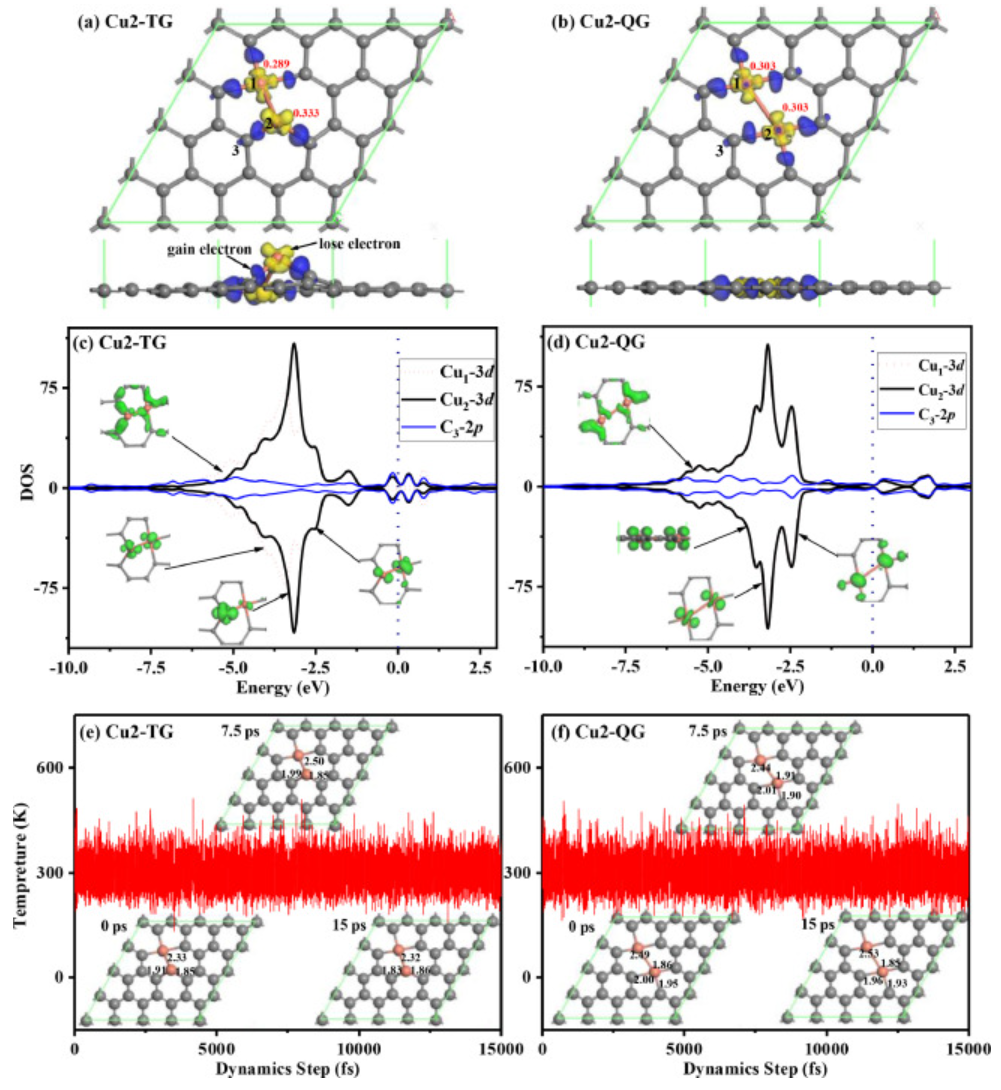

图1. 模型结构、PDOS和AIMD模拟
在具有三个碳原子空位的石墨烯(Cu2-TG)中,一个Cu原子完全位于双空位中,而另一个Cu原子位于由两个C原子和一个Cu元素形成的单空位中,具体如图1a所示。Cu1原子和Cu2原子的吸附能分别为-6.49eV和-3.60eV,这两个吸附值都高于Cu元素的内聚能,即−3.49 eV/atom。因此,Cu2-TG结构可以保持稳定,并且没有团聚问题。在具有四重碳原子空位(Cu2-QG)的石墨烯中,两个Cu原子完全位于两个双空位中,具体如图1b所示。Cu1原子和Cu2原子的吸附能分别为−7.31eV和−5.62eV,这两个吸附值也远高于Cu元素的内聚能。因此,Cu2-QG结构也可以保持稳定,并且有效避免了金属原子的团聚。为了进一步证实Cu2-TG和Cu2-QG的稳定性,作者在300K下用NVT系综进行了15ps的第一性原理分子动力学模拟,具体如图1e和1f所示。作者给出了在0ps、7.5ps和15ps下的三种瞬时结构,从中可以发现Cu2-TG和Cu2-QG的原子位置略有波动,这证实了Cu2-TG、Cu2-QG在室温下可以保持结构动力学稳定性。
Cu2-TG和Cu2-QG中Cu金属原子的差分电荷密度和Hirschfeld原子电荷如图1a和1b所示,其中黄色和蓝色分别表示电子耗尽和电子积累。修饰Cu原子的电子在Cu2-TG和Cu2-QG中都减少了,其中Cu1和Cu2原子将0.333e和0.289e的电子转移到Cu2-TG中的石墨烯,而Cu1和Cu2原子都将0.303e的电子迁移到Cu2-QG中的石墨烯。对于Cu2-TG,空的Cu1–3d轨道平行嵌入石墨烯的基面,而空的Cu2–3d轨道几乎垂直于石墨烯基面。对于Cu2-QG,空的Cu1–3d和Cu2–3d轨道都平行嵌入石墨烯基面中。因此,Cu2-TG的空Cu2–3d轨道具有离域性,并且由于其空间取向,将更容易接受电子,这意味着Cu2-TG对O2和CO分子具有较高的吸附能力。
作者通过PDOS分析发现(图1c和1d),Cu2-TG和Cu2-QG中的Cu和C原子具有较强的相互作用。对于Cu2-TG,C-2p和Cu-3d轨道在−4 eV和−7.5 eV之间显著重叠。而对于Cu2-QG,C-2p和Cu-3d轨道在−4 eV和−7.5 eV之间。对于Cu2-TG,费米能级附近的电子态比Cu2-QG的电子态密度更大,表明Cu2-TG可以将更多的电子转移到分子的空轨道,并促进随后的反应。
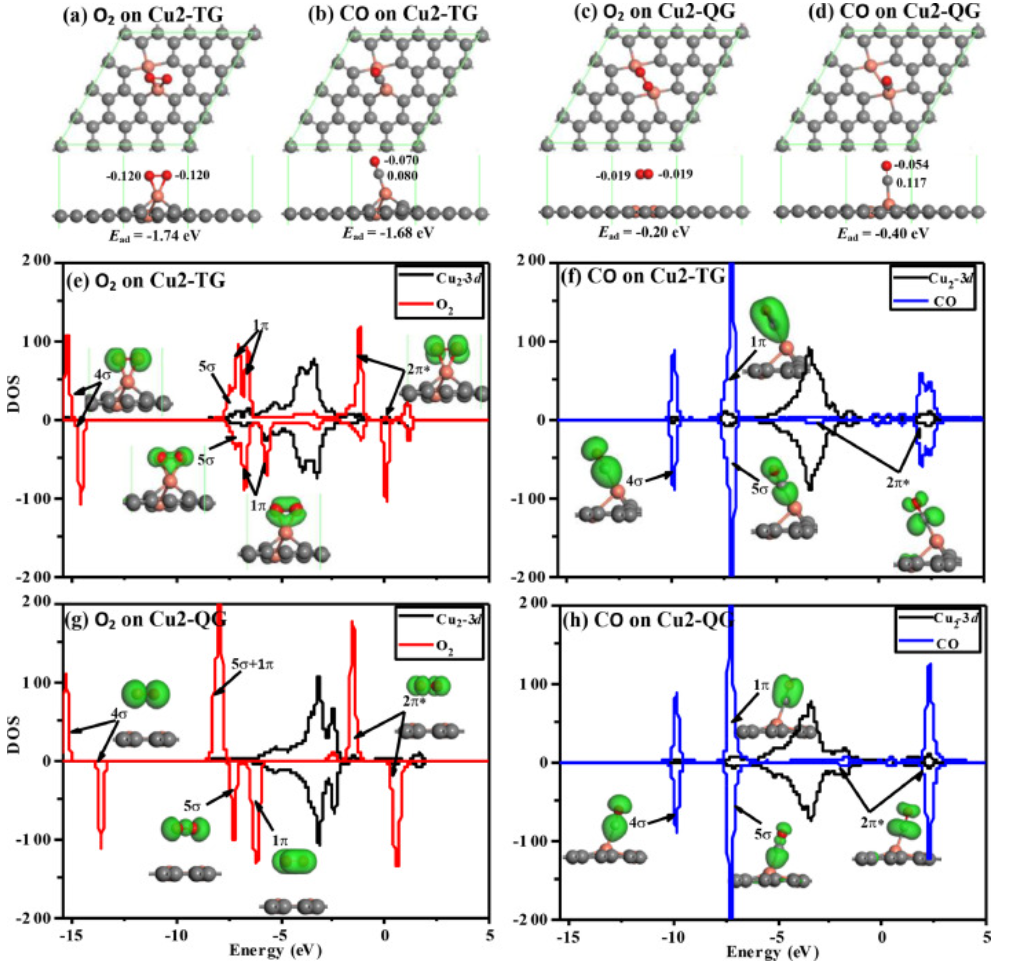
图2. 模型结构和PDOS
在研究CO在Cu双原子催化剂上的氧化之前,作者首先对O2和CO在Cu2-TG和Cu2-QG上的吸附进行了比较研究。图2给出了气体分子在Cu2-TG上的吸附结构,其中O2分子的吸附能为-1.74eV(见图2a),并且O2分子几乎平行于基底。大约0.240e的Hirschfeld电荷从Cu2-TG转移到O2,这导致O-O键从1.225Å延长到1.349Å。吸附的O2通过获得额外的电子而被活化,这将促进O-O键的断裂以及随后的CO氧化。为了确定O2分子的吸附状态,作者计算了O2分子在Cu2-TG上的分裂途径。作者通过过渡态搜索发现,O2分子在Cu2 TG上的分裂能垒为1.75eV,这意味着O2分子不会分裂。图2b显示了CO分子在Cu2-TG上的吸附结构,其中CO分子接近垂直于石墨烯基底,CO和Cu2-TG上的Cu原子之间形成了C-Cu键。在CO吸附后,约0.010e的Hirschfeld电荷从CO转移到Cu2-TG再转移到O2,这导致C-O键从1.141Å延长1.151Å。
相比之下,CO和O2分子在Cu2-QG上的吸附结构如图2c和2d所示,O2和CO分子的吸附能分别为-0.20和-0.40eV。此外,O2在Cu2-QG上的分裂能垒为2.62eV,表明O-O键也不会断裂。作者通过图1c和1d中的PDOS分析发现,Cu2-QG中的Cu1和Cu2原子都有三个Cu-C键,而Cu2-TG中Cu1原子有两个Cu-C键。尽管Cu2-TG中的Cu2原子具有三个Cu-C键,但它在调节Cu1原子的活性方面具有明显的作用,这使得Cu1原子对O2和CO分子具有中等的吸附能。
如图2e所示,Cu-3d轨道将一些电子转移到吸附在Cu2-TG上的O2分子空2π*反键轨道上,而Cu-3d轨道从O2–5σ和O2–1π轨道获得电子。因此,Cu原子可以通过其空3d轨道接受来自O2的电子,并向O2分子提供电子。换句话说,Cu-3d轨道同时具有空轨道和占据轨道,以实现对电子反馈和接受的双重功能,即Cu原子和气体分子之间的电荷转移存在“反馈和接受”机制。相比之下,对于Cu2-QG上的O2分子,在Cu-3d轨道和O2–5σ或O2–1π轨道之间几乎没有电荷转移,而O2–2π*仅从Cu-3d轨道获得少量电子,具体如图2g所示。
如图2f所示,对于吸附在Cu2-TG上的CO分子,Cu-3d轨道将一些电子转移到空CO-2π*反键轨道,即在0~4.0eV范围内形成新的CO峰,而Cu-3d轨道从CO-5σ和CO-1π轨道获得电子,这通过Cu-3d在7.0~7.5eV范围的新峰来证明。尽管CO在Cu2-TG上的吸附过程中也存在“反馈和接受”机制,但CO分子损失的电子比获得的电子多。对于Cu2-QG上的CO,CO-5σ和CO-1π的电子消耗以及CO-2π*轨道的电子增加都受到了抑制,具体如图2h所示。
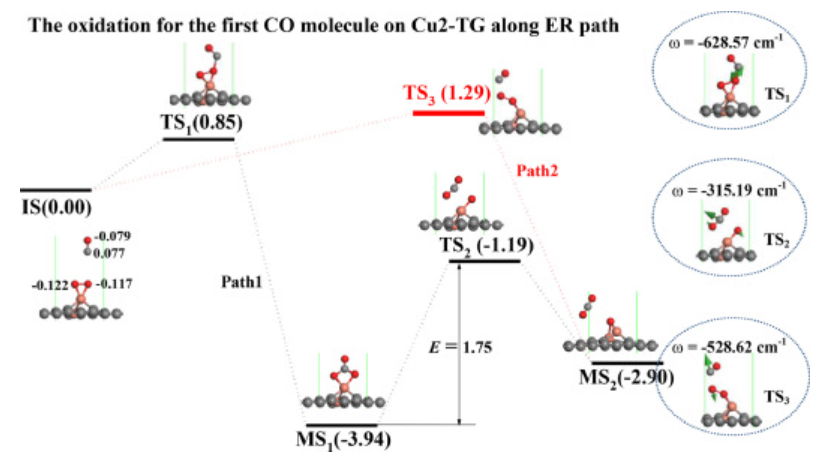
图3. Cu2-TG上CO氧化ER路径反应势能面
为了确定CO分子在Cu2-TG上氧化的最佳反应途径,作者计算了两种ER途径的反应势能面。如图3所示,对于ER-路径1的反应过程,作者选择在Cu2-TG上具有悬浮CO分子和预吸附O2分子的结构作为初始状态,当CO攻击O2分子时,会产生新的O-C键来连接CO和O2分子(见图3中的TS1)。在通过0.85eV的反应能垒后,O2分子分裂成两个O原子,并且CO分子吸附在两个O上,从而在Cu2-TG上形成CO3中间体(见图3中的MS1),该过程释放3.94eV的热量。随后,在Cu2-TG上越过1.75eV的反应能垒后形成第一个CO2分子(见图3中的MS2)。对于ER-路径2,在Cu2 TG上越过1.29eV的反应能垒后产生CO2产物(见图3的TS3)。
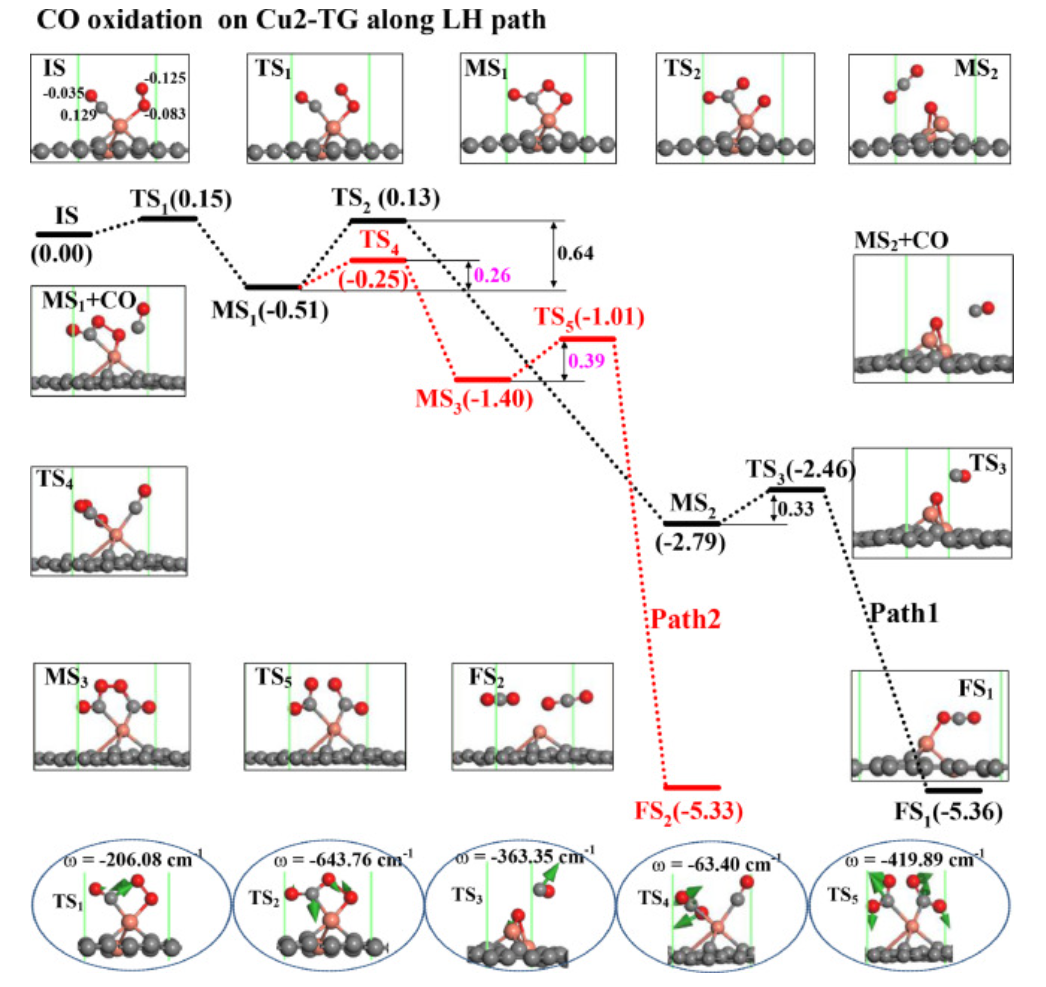
图4. Cu2-TG上CO氧化LH路径势能面及模型结构
对于LH路径(图4),作者采用共吸附在Cu2-TG上的CO和O2分子的结构作为初始状态(见图4中的IS),其在越过0.15eV的能垒后,形成由OCOO中间体和Cu2-TG组成的MS1结构。对于OCOO中间体的后续反应,有两种可能的途径,即OCOO中间体离解成一个CO2分子和一个O原子(见图4中的途径1),或者OCOO中间体与另一个CO分子相互作用形成OCOOCO中间体(见图4的途径2)。
沿着LH路径1,第一个CO2分子在Cu2-TG上产生,其能垒为0.64eV(见图4中的MS2),而MS1中OCOO中间体的O-O键在TS2处分裂,其中CO2分子在TS2的键角为136.1°。随后,CO分子将与源自O2分子的第二个O原子在越过0.33eV的反应能垒后形成第二个CO2分子。LH路径1之后的步骤3释放2.57eV的热量,这远高于第二个CO2在Cu2-TG上的吸附能(-0.37eV),并且可以提供足够的能量使CO2分子从Cu2-TG表面分离。因此,沿LH路径1的速率限制能垒为0.64eV。
对于LH路径2,OOCO中间体与另一个CO(见图4中的MS1+CO)相互作用,在通过0.26eV的反应能垒后形成OCOOCO中间体(见图4的MS3)。然后,在TS5处越过0.39eV的反应能垒后,在Cu2-TG上形成两个CO2分子。OCOOCO结构在MS3处的O-O键在TS5处分裂,两个变形的CO2分子吸附在Cu原子上,该反应步骤释放3.93eV的热量,其远大于CO2在Cu2-TG上的吸附能。
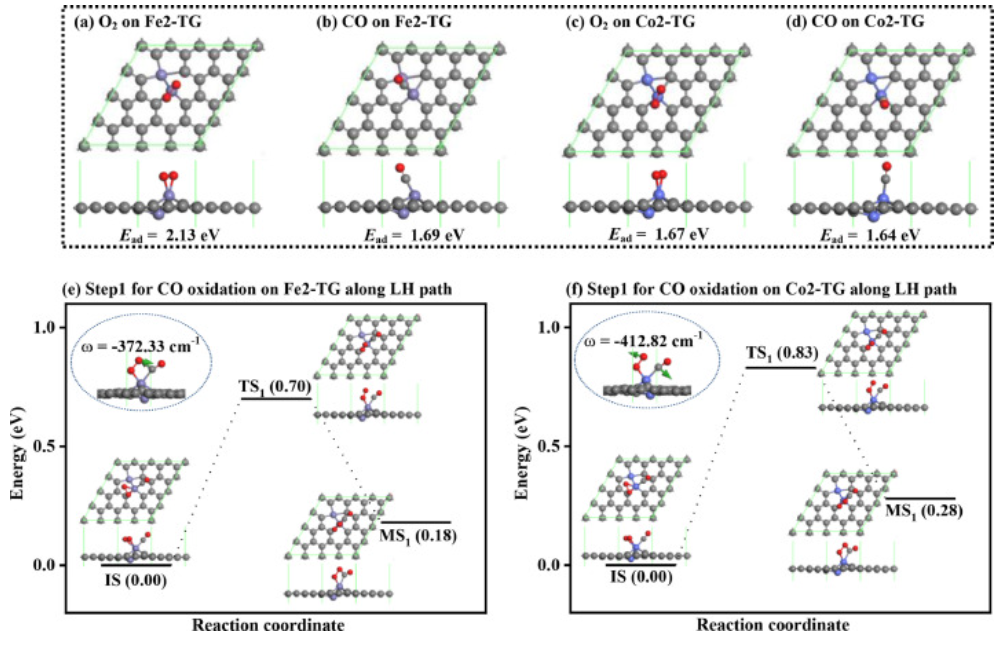
图5. 气体分子吸附构型和沿LH路径进行CO氧化的第一步势能面
O2和CO分子在Fe2-TG和Co2-TG上的吸附结构和吸附能计算如图5a–5d所示,作者计算了沿LH路径在Fe2-TG和Co2-TG上进行CO氧化的第一步以评估它们对CO氧化的性能。如图5e和5f所示,Fe2-TG和Co2-TG上LH路径的第一步反应能垒分别为0.70eV和0.83eV,这远大于Cu2-TG的反应势垒。此外,这两个反应步骤都是吸热的,不利于反应的自发进行。因此,在CO氧化过程中,Cu2-TG比Fe2-TG和Co2-TG具有更好的性能。
结论与展望
在Cu2-TG中,通过从Cu原子到碳原子的电荷转移产生了空Cu-3d轨道,该轨道对于一个Cu原子平行于石墨烯基面,而对于另一个铜原子几乎垂直于石墨烯基面。而对于Cu2-QG中的两个Cu原子,空的Cu-3d轨道平行于石墨烯基面。由于Cu2-QG与气体分子之间的弱相互作用,CO沿ER路径的最大氧化能垒为1.75eV,而在LH路径上仅为0.39eV。电子结构表明,铜原子可以通过其空3d轨道接受电子,并通过其填充的3d轨道失去电子,从而具有“接受和反馈”机制。这些结果表明,三碳空位使Cu2TG成为一种有效的CO氧化催化剂。
文献信息
Quanguo Jiang et.al Effects of carbon vacancies on the CO oxidation on Cu double atom catalyst supported by graphene Surfaces and Interfaces 2023,https://doi.org/10.1016/j.surfin.2023.103312
本文来自催化开天地,本文观点不代表石墨烯网立场,转载请联系原作者。