
氧还原反应(ORR)在不同的能源和可持续性领域仍然处于研究的前沿。虽然石墨烯负载的单原子催化剂(SAC)因优化ORR效率而备受关注,但调整相邻单原子位点之间的相互作用带来了复杂的挑战。在这项研究中,本文利用密度泛函理论(DFT)计算和尖端的机器学习(ML)技术探索了144个石墨烯支持的SAC,其特征是相互作用的M1–N4和M2–N4部分(M1,M2=Mn、Fe、Co、Ni、Cu、Ru、Rh、Pd、Ag-Ir、Pt、Au),表示为M1–M2。通过调整这些相互作用,本文发现了13种优于基准催化剂Fe(OH)–N4的特殊SAC,包括性能最好的Fe–Pd和几种非贵金属SAC,如Fe–Ag、Ag–Cu和Ag–Ag。进一步探索,本文的ML模型有效地捕捉了单原子金属性质和过电位之间的相关性,为合理的电催化剂设计提供了工具。我们的研究阐明了高效SAC催化ORR的道路,促进了可持续、节能的未来。
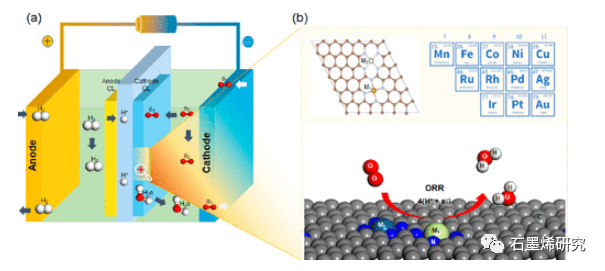
图1. (a)原电池反应和(b)M1–M2 SAC上的ORR的示意图。
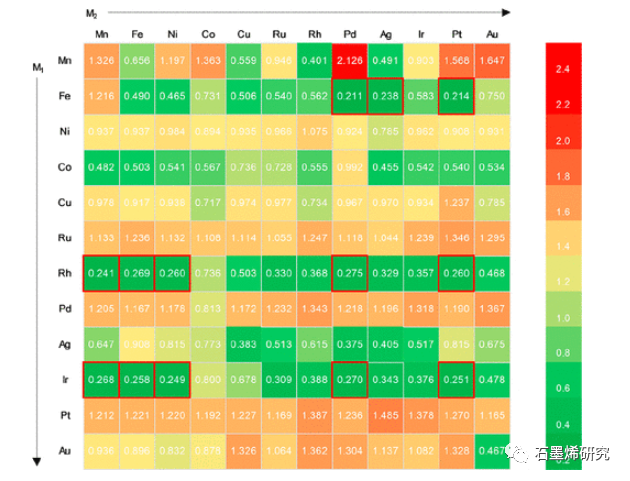
图2. 144个SAC的过电位。绿色值表示低过电位(高活性),而橙色/红色值表示高过电位(低活性)。η<0.304 V的有希望的SAC用红色方块突出显示。
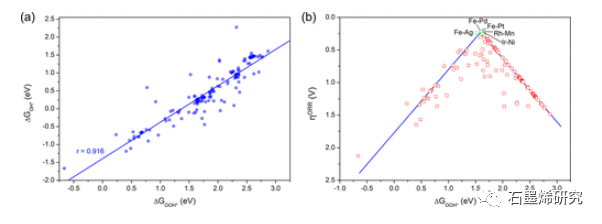
图3. (a) 144个SAC的ΔGOOH*和ΔGOH*之间的关系图。(b) 144个SAC的过电位作为ΔGOOH*的函数。表现最好的五个M1–M2 SAC被涂成绿色并贴上标签。火山图的顶点强调了ΔGOOH*(~1.59–1.78 eV)的最佳结合能范围。
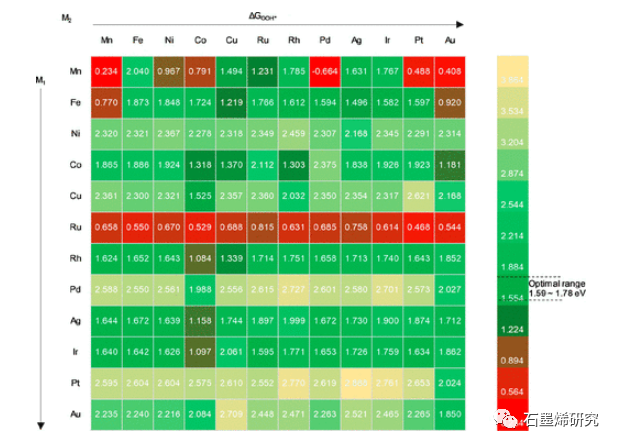
图4. ΔGOOH*用于144个SAC。最佳结合能(~1.59–1.78 eV)用绿色表示,过强结合能用红色表示,过弱结合能用黄色表示。
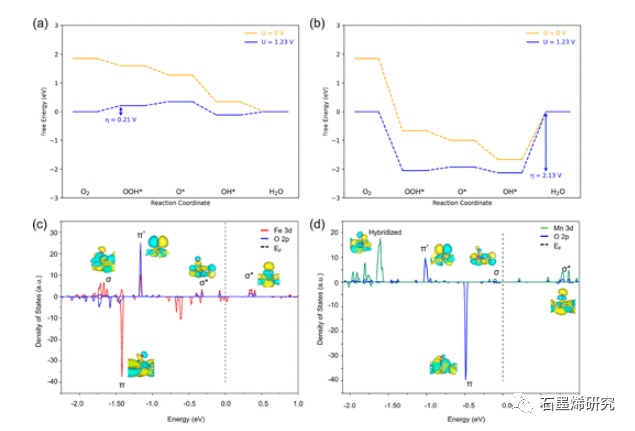
图5. (a)性能最好的SAC(Fe–Pd)和(b)性能最差的SAC的自由能分布的比较。OH*结合后(c)Fe–Pd和(d)Mn–Pd的计算PDOS。成键分子轨道,表示为σ和π,对应于Fe或Mn的d轨道与O的p轨道之间的成键相互作用。
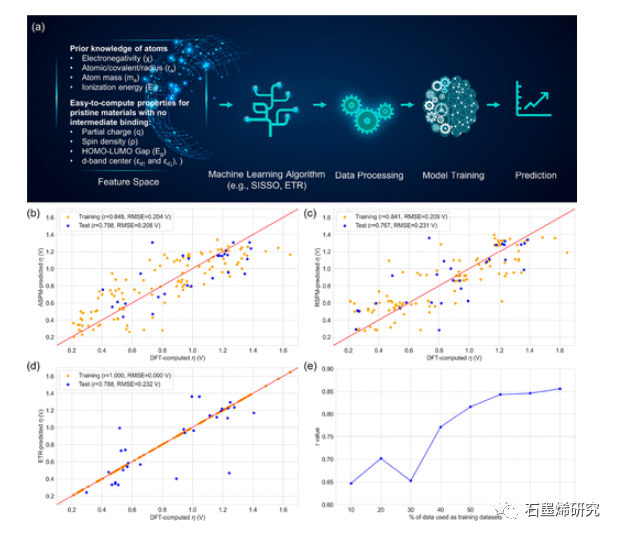
图6. (a) 根据本征原子性质和DFT计算的电子性质预测过电势的ML过程的工作流程。训练(80%)和测试(20%)数据集的(b)ASPM-SISSO、(c)RSPM-SSISSO和(d)ETR模型的预测性能。(e) 说明随着训练数据集大小的增加,ETR模型的预测能力逐渐提高。
相关研究成果由美国西北大学Xijun Wang等人2023年发表在The Journal of Physical Chemistry Letters (翻译:https://doi.org/10.1021/acs.jpclett.3c02273)上。原文:Fine-Tuning Dual Single-Atom Metal Sites on Graphene toward Enhanced Oxygen Reduction Reaction Activity。
本文来自石墨烯研究,本文观点不代表石墨烯网立场,转载请联系原作者。