
研究背景
基于可调带隙的多组分二维(2D)过渡金属二卤代化合物(TMDCs)半导体被越来越多地应用于设计具有特定光谱响应的光电器件。但是在理论层次对其进行分析依旧缺少完善的理论支撑。
中国人民大学王志永等人采用合金化和多相复合的结合思路,设计了带隙可调的MoxW1-xS2/石墨烯异质结构。从理论上研究了MoxW1-xS2/石墨烯异质结的接触类型、稳定性和光电性能。
计算方法
所有的计算都是基于DFT下的DMol3和CASTEP软件包来实现的。在DMol3中,采用广义梯度近似(GGA)下的Perdew-Burke-Ernzerhof (PBE)泛函描述电子交换相关相互作用。
布里渊区K点设置为9×9×1,直到最大力收敛精度为3×10-2 eV/Å。DMol3和CASTEP都采用特卡琴科-谢弗勒(TS) 方法来进行分子间色散校正,并对所有系统应用15 Å真空层,以防止同一研究对象之间的层间相互作用。
结果与讨论
可以看出,单分子层MoS2、WS2和石墨烯的优化晶格参数分别为3.169、3.192和2.465 Å,与之前报道的结果非常接近。在此基础上,选择2×2×1单层二硫化钼和WS2构建单层Mo0.25W0.75S2和Mo0.75W0.25S2合金模型(图1a-d),并对其进行几何优化。
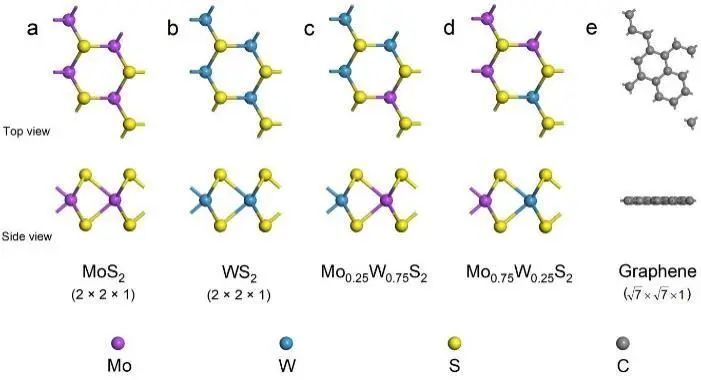

图1. 催化剂结构图
Mo0.25W0.75S2和Mo0.75W0.25S2的不同匹配模型如图2所示。通过计算异质结在平衡层间距处的形成能,发现对于Mo0.25W0.75S2/G(G为石墨烯,以下相同),模式(5)的形成能最低;在Mo0.75W0.25S2/G的情况下,模式(2)的形成能最低。因此,我们选择(5) Mo0.25W0.75S2/G和(2) Mo0.75W0.25S2/G作为本研究的初步研究对象。
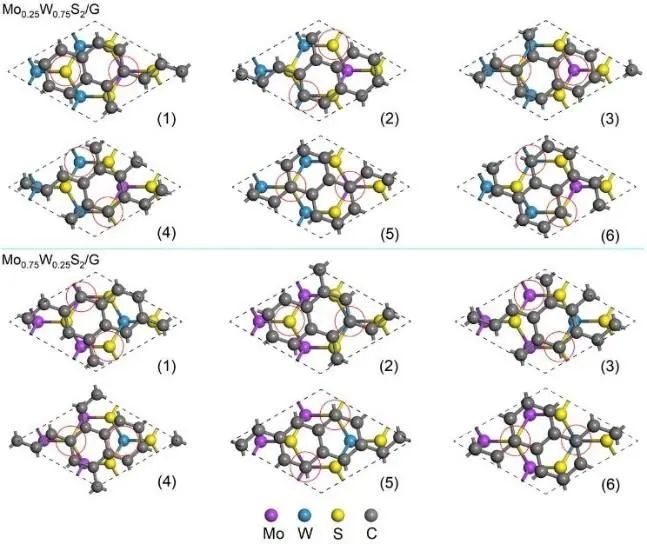
为了进一步说明系统的稳定性,作者还进行了从头算分子动力学(Ab initio molecular dynamics, AIMD)模拟,在室温为300 K时,Mo0.25W0.75S2/G和Mo0.75W0.25S2/G均保持异质结构的完整性,在模拟结束时没有明显变形,时间步长为1 fs,持续时间为5 ps,两种异质结构的总能量保持在平均值,基本保持不变,说明两种体系在室温下是稳定的。
确定初始结构后,图3显示了异质结形成能与层间距的关系。通过调整MoxW1-xS2与石墨烯的层间距,发现Mo0.25W0.75S2/G和Mo0.75W0.25S2/G的形成能在层间距分别为3.372 Å和3.373 Å时最低。这些距离非常接近MoS2/石墨烯异质结的最佳层间距(3.4 ű0.1 Å)。.这些值也在典型的范德瓦尔斯结合能范围内,因此MoxW1-xS2/G异质结构是一个范德瓦尔斯异质结。
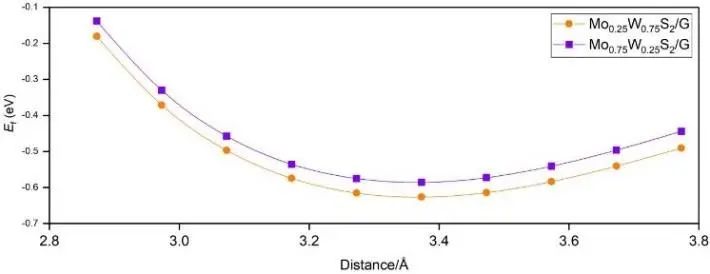
图3. 异质结层间距与形成能的关系
为了研究MoxW1-xS2/G异质结在光电领域的应用,必须对整个异质结及其组成部分的光电性能进行研究。我们首先计算了单层MoxW1-xS2、石墨烯和MoxW1-xS2/G的能带结构。
为了便于比较,还计算了本征单层MoS2和WS2的带隙。结果如表1和图4所示(图4中所有系统的费米能级都设置在0 eV位置,并用虚线标出)。可以看出,单层Mo0.25W0.75S2和Mo0.75W0.25S2的导带最小值(CBM)和价带最大值(VBM)分别对应于布里渊带的高对称点K,表现出直接带隙的特性,这对基于该材料制备光电器件非常有利。
与本征单层MoS2的带隙值相比,Mo0.25W0.75S2和Mo0.75W0.25S2的带隙值分别为1.877 eV和1.791 eV,这与也反映了当Mo元素在单层MoxW1- xS2中占一定浓度时,带隙相对于本征单层MoS2不增反减的趋势。
值得注意的是,石墨烯在两种异质结构中分别打开了约0.735和0.844 meV的小带隙。这些小带隙是石墨烯堆叠成异质结构后晶格对称性被破坏的结果。
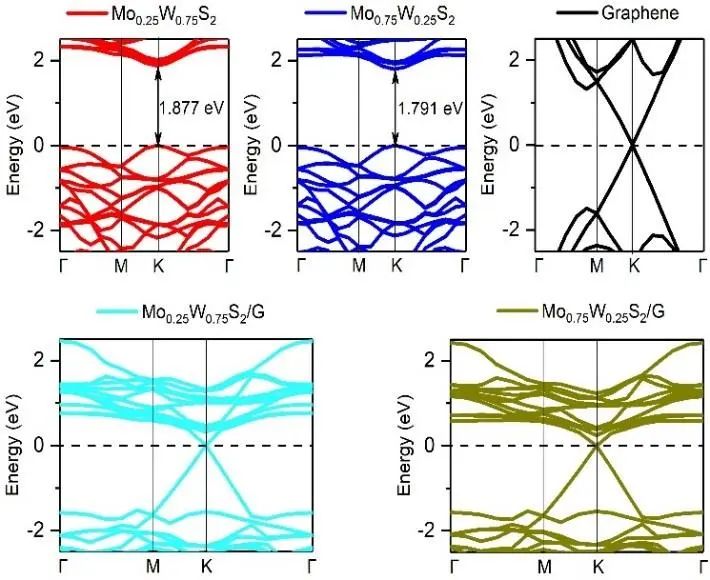
图4. 异质结构能带
当两组分材料接触形成异质结时,界面效应可能导致两层材料之间的电荷转移。因此,我们计算了MoxW1-xS2/G的电子密度差。图5a为Mo0.25W0.75S2/G和Mo0.75W0.25S2/G异质结在其平衡距离处的电子密度差,其中红色区域表示电子减少,蓝色区域表示电子增加。
显然,石墨烯在与MoxW1-xS2接触时充当电子供体,电子在MoxW1-xS2层聚集,在石墨烯层消散。当石墨烯与MoxW1-xS2形成异质结时,石墨烯上的电子被转移到MoxW1-xS2上,导致MoxW1-xS2表面积累负电荷,石墨烯表面积累正电荷,从而在界面处形成内置电场。
MoxW1-xS2中S原子附近的光生电子倾向于向石墨烯转移,这有利于光生电子-空穴对的有效分离。这对提高MoxW1-xS2的光催化性能具有重要意义。特别需要指出的是,当MoS2 (WS2)接触石墨烯时,光生电子会通过内置电场从MoS2 (WS2)转移到石墨烯上,这一点得到了实验和理论计算的证实。
为了探讨MoxW1-xS2/G在光催化水裂解析氢及析氧中的可能性,作者比较了MoxW1-xS2/G与水裂解氧化还原电位的相对位置。如图5b所示,为了实现光催化剂对水的分解,需要将导带和价带的边缘与水的还原和氧化电位相匹配。
即CBM小于水的还原电位(0 eV vs. NHE, pH=0),而VBM大于水的氧化电位(1.23 eV vs. NHE, pH=0),带隙应大于水的劈裂电压。此时,光辐射可以有效地使电子从价带跃迁到导带。
由图5b可以看出,MoxW1-xS2/G可以满足劈水电化学的要求。MoxW1-xS2/G的CBM小于水裂解还原电位,而VBM大于水裂解氧化电位。导带和价带的边缘位置与水的还原电位和氧化电位相匹配,说明光生电子和空穴可以在MoxW1-xS2/G上发生氧化还原反应,进而析出氢气和氧气。
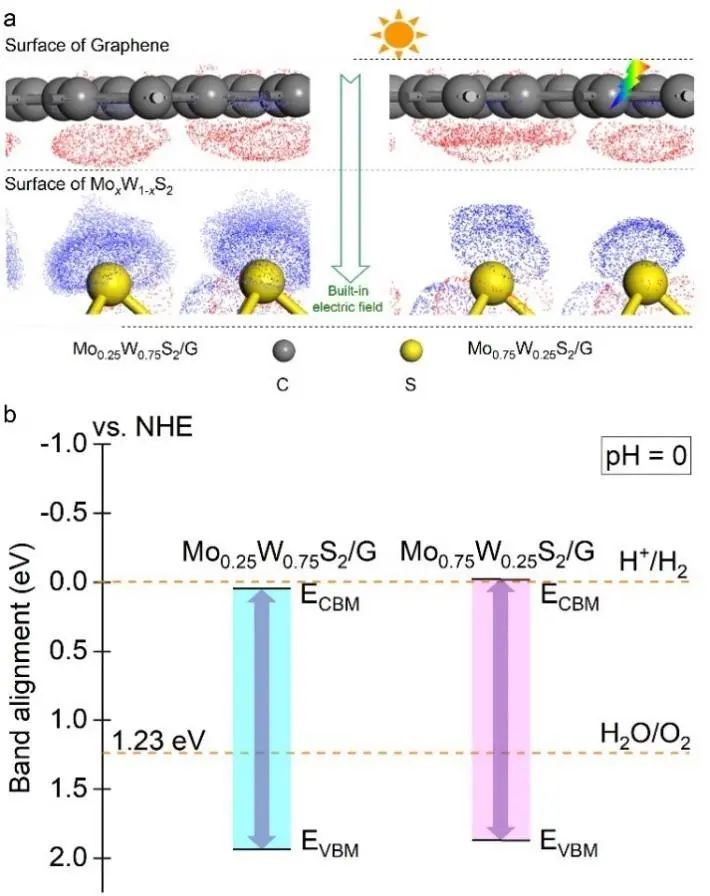
图5. 异质结差分电荷及带边位置
当MoxW1-xS2与石墨烯形成异质结构时,可能形成肖特基接触或欧姆接触,如图6所示。从图4的能带结构可以看出,Mo0.25W0.75S2/G和Mo0.25W0.75S2/G在平衡距离处都是肖特基触点。
根据Schottky- Mott规则,Mo0.25W0.75S2/G的n型肖特基势垒(ΦB, n)和p型肖特基势垒(ΦB, p)的高度分别可计算为0.318 eV和1.557 eV;Mo0.25W0.75S2/G的n型肖特基势垒高度为0.245 eV, p型肖特基势垒高度为1.541 eV。有报道称,电场可以有效地控制范德华异质结的SBH,实现异质结从n型肖特基接触向p型肖特基接触的转变。这种通过施加电场改变触点类型的方法有望扩大异质结在肖特基器件中的应用。
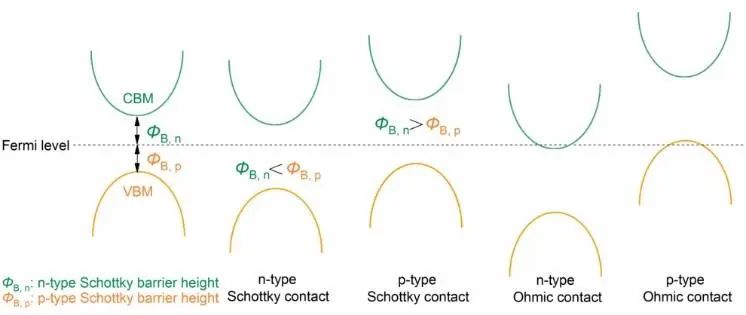
图6. 金属-半导体异质结的接触类型示意图
结论与展望
本文采用DFT计算方法研究了MoxW1-xS2/G异质结的光电性能。结果表明,Mo0.25W0.75S2和Mo0.75W0.25S2都是具有直接带隙的半导体。研究表明本征单层MoS2和WS2对太阳光的吸收范围可以通过合金化来调节,以提高其作为光电器件的应用价值。
内置电场的存在促进了MoxW1-xS2光生电子空穴对的有效分离。此外,施加垂直外电场,可以动态调节MoxW1-xS2与石墨烯的接触类型,实现从n型肖特基接触向p型肖特基接触的转变。为基于MoxW1-xS2/G的光电器件的实验设计提供理论参考。
文献信息
Chen, J., Zhou, Z., Li, Z., & Wang, Z. (2023). Theoretical design of MoxW1− xS2/graphene heterojunction with adjustable band gap: potential candidate materials for next generation of optoelectronic devices. ChemPhysChem, e202300095.
https://doi.org/10.1002/cphc.202300095
本文来自催化开天地,本文观点不代表石墨烯网立场,转载请联系原作者。