
基于第一性原理的密度泛函理论,研究了合金原子在铝基体和石墨烯/铝界面中的扩散迁移行为。通过对比在铝基体和石墨烯/铝界面上铝原子的迁移势垒,发现铝原子更倾向于向界面迁移,为脆性相Al4C3的形成提供了条件。计算了41个合金原子在铝基体和石墨烯/铝界面处的扩散迁移行为,选择了一组在界面处倾向于聚集的合金元素(Sc、Cu、Si、Ni等)。还发现Si、Cu、Ni和Co原子比Al原子更容易迁移到界面上。最后,研究了具有碳原子空位缺陷的石墨烯/铝界面上合金原子的迁移行为,与完美界面相比,大多数合金原子在具有碳原子空位缺陷的石墨烯/铝界面上的迁移能变化不大,而Sc和B原子更倾向于具有空位缺陷的石墨烯/铝界面。理论研究结果有望为未来高性能石墨烯/铝复合材料的实验设计提供理论依据。
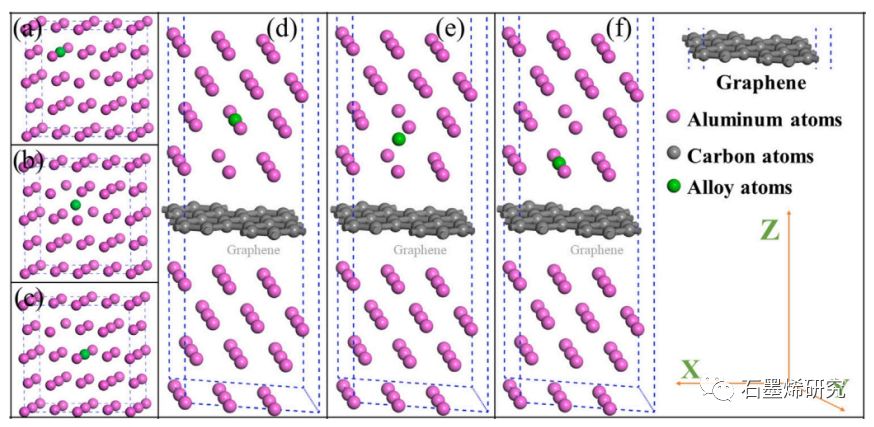

图1. 合金原子在铝基体和界面中扩散迁移的晶体结构,其中紫色原子为Al原子,灰色原子为C原子,青色原子为掺杂合金原子。
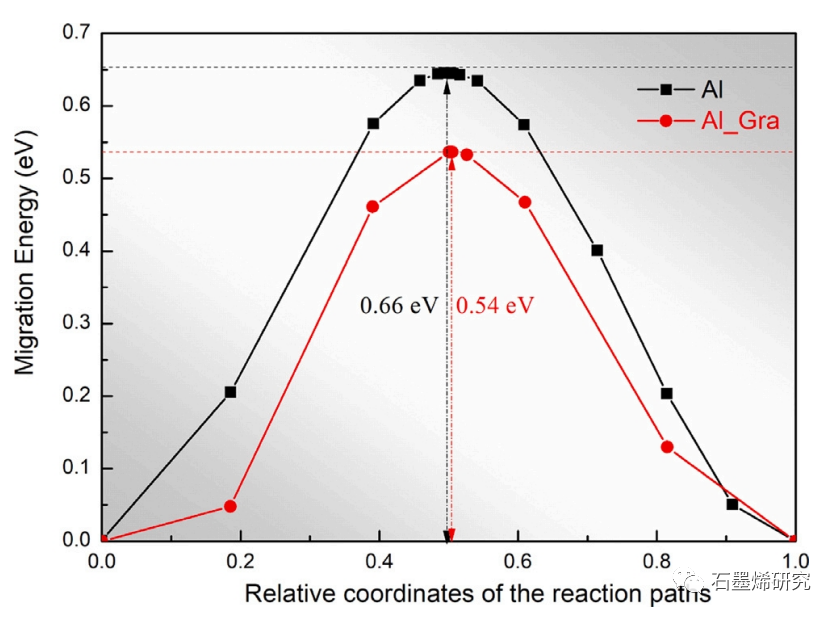
图2. 铝原子在铝基体和石墨烯/铝复合材料界面的迁移能。
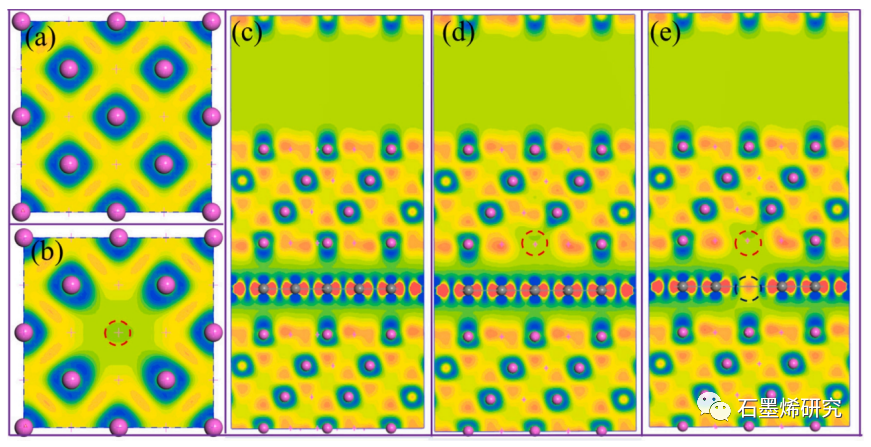
图3. (a)完美铝、(b)有缺陷铝晶体、(c)石墨烯/铝界面、(d)缺陷石墨烯/铝界面、(e)缺陷石墨烯/合金原子/铝界面的电荷密度差。
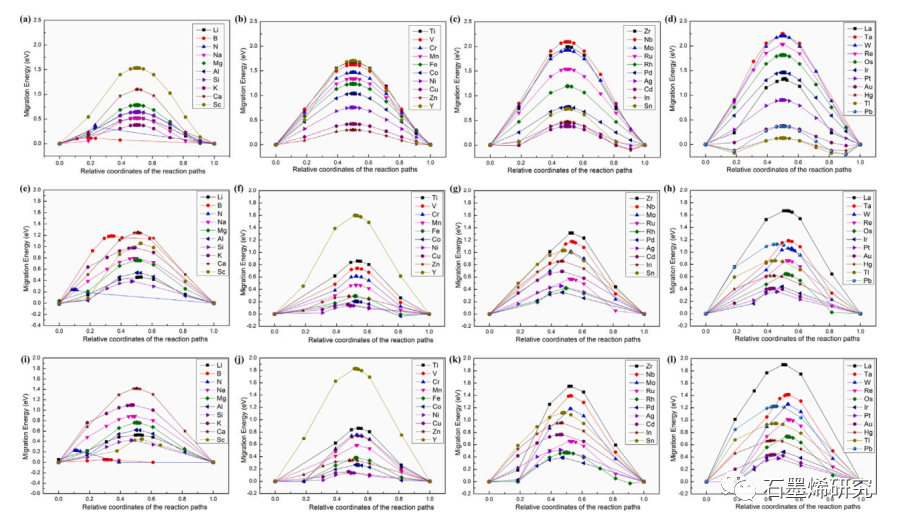
图4. Li, B, N, Na, Mg, Al, Si, K, Ca, Sc;Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Y;Zr, Nb, Mo, Ru, Rh, Pb, Ag, Cd, In, Sn;La, Ta, W, Re, Os, Ir, Pt, Au, Hg, Tl, Pb合金原子在(a)铝基质中、(b)石墨烯/铝界面、(c)有碳原子空位缺陷的石墨烯/铝界面处的迁移能量。
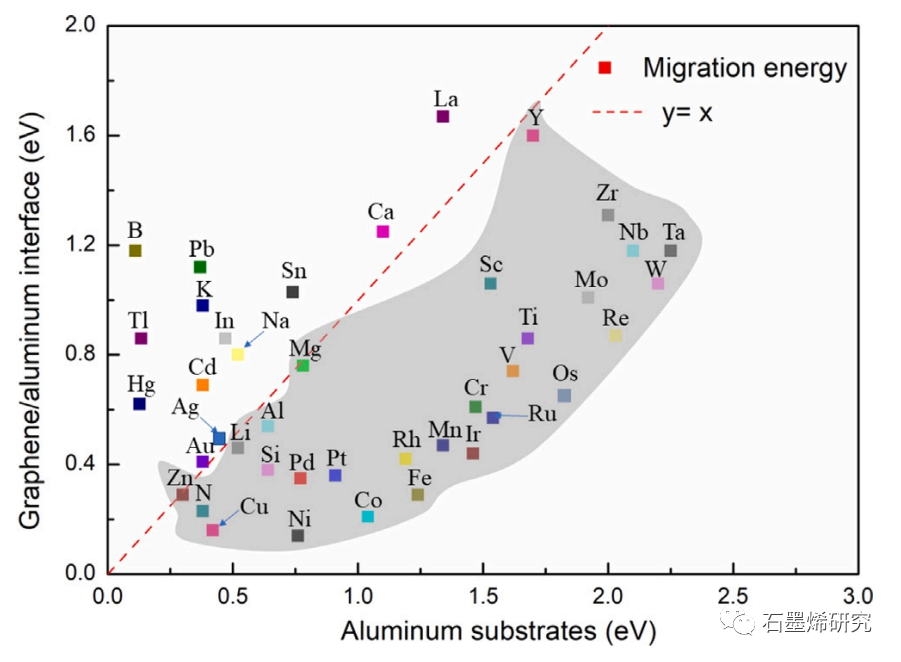
图5. 合金原子在铝基体和石墨烯/铝界面上迁移能的比较。
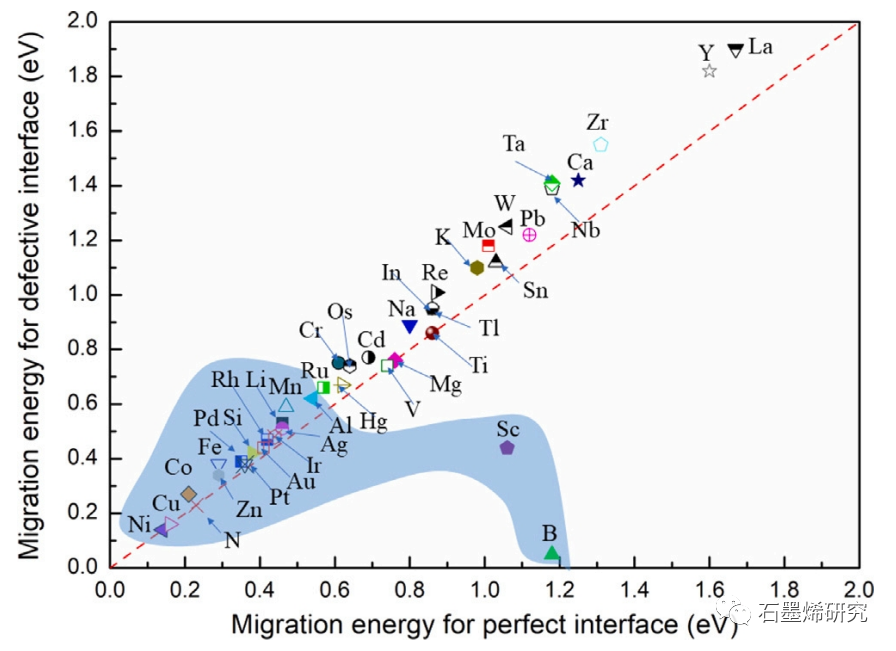
图6. 在完美石墨烯/铝界面与缺陷石墨烯/铝界面合金处原子迁移能的比较。
相关研究成果由哈尔滨工业大学材料科学与工程学院 Jingchuan Zhu等于2023年发表在Surfaces and Interfaces (https://doi.org/10.1016/j.surfin.2023.102825)上。原文:Insights into the diffusion migration behavior of alloy atoms at the graphene/aluminum interface: First-principles calculations。
本文来自石墨烯研究,本文观点不代表石墨烯网立场,转载请联系原作者。