
采用密度泛函理论(DFT)计算方法研究了官能团(羟基和环氧基)对氧化石墨烯与氧化铁基体界面相互作用的影响。计算结果表明,吸附的官能团可以提高GO与Fe2O3基底之间的相互作用强度,结合能增强的幅度与官能团的吸附位有关。对于位于表面O原子顶部氧化原子相邻C原子的GO/ Fe2O3-O模型,结合能显著增强,这主要是由于界面C-O共价键的形成。此外,GO/ Fe2O3-O模型的最大结合能是Gr/ Fe2O3-O模型的4倍以上。对于本文研究的GO/Fe2O3-O和GO/Fe2O3-Fe模型,GO在Fe2O3表面发生物理吸附,结合能略有提高。该研究有助于更深入地了解石墨烯基涂层材料在氧化铁表面的防护性能。
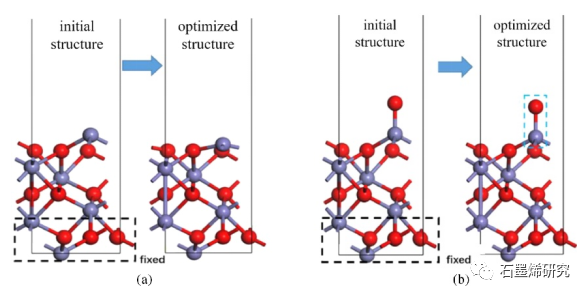

图1. 研究了(a)Fe2O3-Fe和(b)Fe2O3-O表面的初始结构和优化结构。氧化铁的铁原子和氧原子分别用紫色和红色表示。
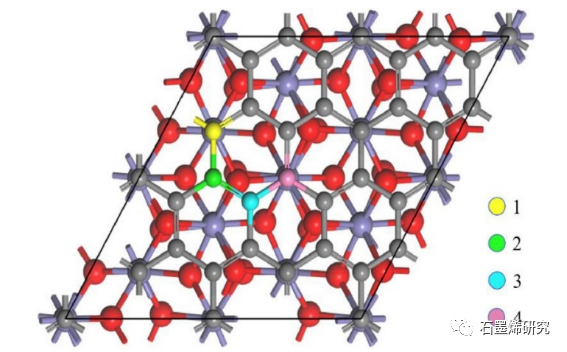
图2. Gr/Fe2O3-Fe模型中石墨烯层上四个具有代表性的官能团吸附点。C原子、Fe原子和O原子分别显示为灰色、紫色和红色。吸附位点1、2、3和4上的C原子分别用黄色、绿色、蓝色和粉色标记。
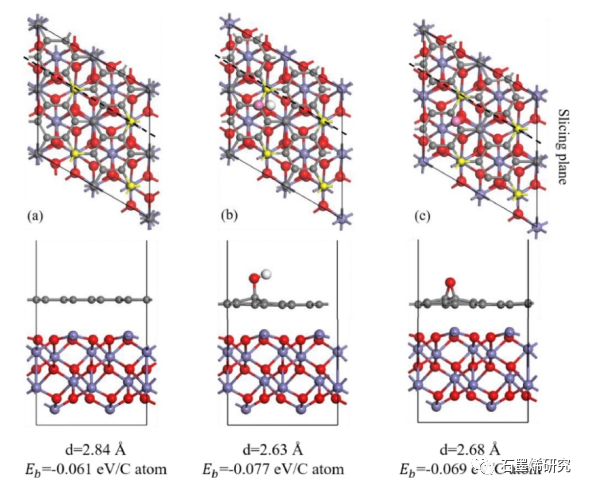
图3. (a)Gr/Fe2O3-Fe,(b)GO-OH-2/Fe2O3-Fe,(c)GO-O-23/Fe2O3-Fe模型优化结构的垂直视图(上)和横向视图(下)。垂直视图中,Fe原子表面顶部的C原子用黄色标记,官能团的O原子用粉色标记。
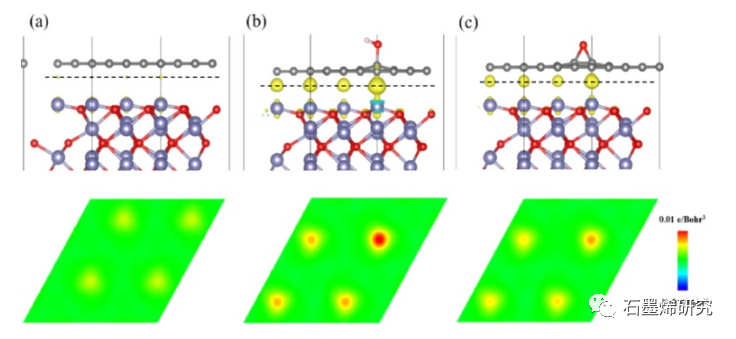
图4. (a)Gr/Fe2O3-Fe,(b)GO-OH-2/Fe2O3-Fe,(c)GO-O-23/Fe2O3-Fe模型的电荷密度差(上)及其二维图(下)。上图中的Fe、O、C和H原子分别以蓝色、红色、灰色和白色显示。青色和黄色表示电子的消耗和积累,等值面值为0.002 e/Bohr3。
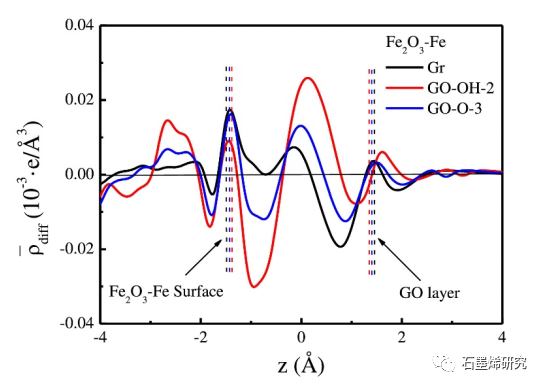
图5. Gr/Fe2O3-Fe、GO-OH-2/Fe2O3-Fe和GO-O-23/Fe2O3-Fe模型沿z-方向面平均电荷密度差。左(右)虚线之间的空间表示Fe2O3-Fe表面和GO层之间的界面。
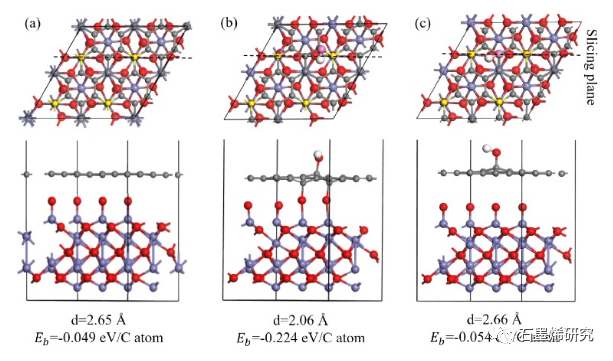
图6. (a)Gr/Fe2O3-O,(b)GO-OH-2/Fe2O3-O,(c)GO-OH-3/Fe2O3-O模型优化结构的垂直视图(上)和横向视图(下)。在垂直视图中,O原子表面顶部的C原子用黄色标记,官能团中的O原子用粉色标记。
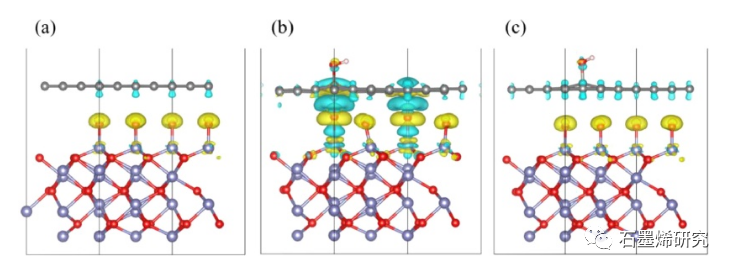
图7. (a)Gr/Fe2O3-O,(b)GO-OH-2/Fe2O3-O,(c)GO-OH-3/Fe2O3-O模型的电荷密度差。青色和黄色表示电子的消耗和积累,等值面值为0.003 e/Bohr3, Fe、O、C和H原子分别显示为蓝色、红色、灰色和白色。
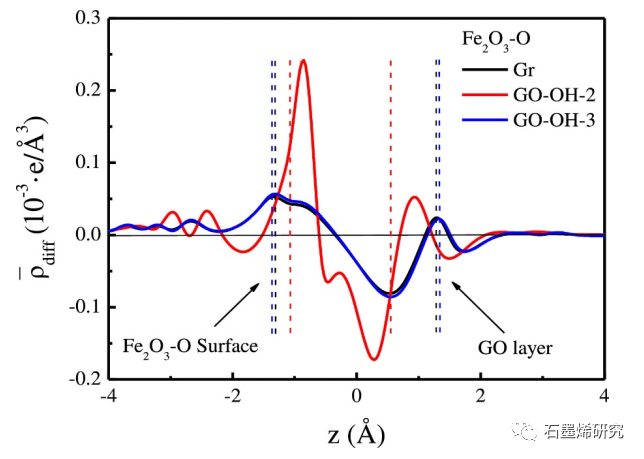
图8. Gr/Fe2O3-O,GO-OH-2/Fe2O3-O,GO-OH-3/Fe2O3-O模型沿z-方向面平均电荷密度差。左(右)虚线之间的空间表示Fe2O3-Fe表面和GO层之间的界面。
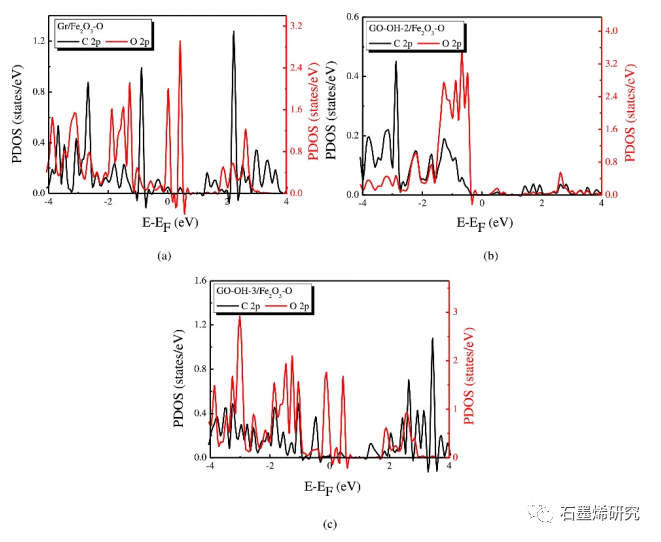
图9. Gr/Fe2O3-O,GO-OH-2/Fe2O3-O,GO-OH-3/Fe2O3-O模型中顶部C原子2p轨道和O原子下方2p轨道的PDOS。
相关研究成果由重庆大学机械传动国家重点实验室Xia Wang等人于2021年发表在Surface Science (https://doi.org/10.1016/j.susc.2021.121982)上。原文:Effect of functional groups on the adsorption of graphene oxide on iron oxide surface。
本文来自石墨烯研究,本文观点不代表石墨烯网立场,转载请联系原作者。