
英文原题:Quantum Tunnelling Driven H₂ formation on Graphene


通讯作者:陈基,北京大学
作者:韩尔逊,方为,Michail Stamatakis,Jeremy O. Richardson
研究背景
氢作为最轻的元素,其原子核的量子隧穿在许多物理,化学以及生物反应过程中发挥着重要作用。虽然氢元素是最轻的元素,但是氢却占据了宇宙总质量的75%。尤其是在太空环境中,氢的丰度及其演化是宇宙化学过程的核心。氢的演化过程中最基本的过程就是两个氢原子复合成一个氢分子的过程,没有这个过程就没有氢气的存在。氢原子随机碰撞复合成氢气分子的过程是一种最直观的路径,但是近些年的研究表明仅仅通过随机碰撞过程导致的氢气含量是远低于真实情况的,所以一定存在某种异质的催化过程来加速氢气分子的复合。其中,碳物质表面被认为是一种天然存在的高效的氢气复合的催化剂。然而,经典的理论,不考虑氢原子核的量子效应,所预测的碳物质表面的氢复合速率仍然非常低,尤其是在低温下,远远没有达到能解释太空中氢气浓度的预期反应速率。另一方面,有越来越多的关于石墨烯等碳材料的低温真空实验也需要理论上加深到对吸附氢及其反应过程的微观理解。
快讯亮点
本工作结合最新发展的第一性原理瞬子理论和动力学蒙特卡洛模拟,研究了石墨烯和其他碳物质表面氢原子的吸附、扩散和复合过程:
(1) 精确计算了不同温度下氢原子复合、扩散以及脱附的量子隧穿速率;
(2) 发现了在经典情况下被禁止的氢复合路径在考虑量子隧穿后变成重要过程;
(3) 深入分析了多维势能面上经典反应路径和量子反应路径的区别,揭示了反应过程多维隧穿效应和量子角切效应。
内容介绍
在化学反应速率的经典描述中,反应速率随着反应所需要跨越的经典能垒以温度的倒数呈指数衰减。而量子隧穿会使得低温下的反应速率趋近于一个有限的常数,从而使得在经典情况下被完全抑制的反应能够以可观的速率发生。经典速率和量子速率的典型行为我们在后文具体展示。总之,低温下反应速率的准确计算离不开对于量子隧穿效应的处理。路径积分瞬子理论(ring-polymer instanton theory)是一种通过寻找最优隧穿路径计算隧穿速率的新方法,目前在气相反应中有较广泛的应用。这种方法兼顾准确性与计算效率,在表面体系与多维凝聚态系统中也拥有广泛的应用前景。本工作中将第一性原理电子结构计算和瞬子理论进行实时的结合,可以用来更准确的探究多维势能面上的量子隧穿路径和计算反应速率。
图1中展示了典型的氢的复合、扩散和脱附过程。图2(左)给出了这些过程对应的反应速率随着温度的变化关系。其中实线和虚线分别对应量子和经典的反应速率。可以看到,如果没有量子隧穿,氢的复合过程的经典的反应速率在100K以下就衰减到可以忽略不计的量级。而考虑了量子隧穿,氢在不同位置的复合过程都有一定可观的速率,相比于经典速率有几十个数量级以上的提高。图2右则是综合所有反应过程的速率进行的动力学蒙特卡洛模拟所得到的各个过程的发生频率。通过经典与量子的对比,可以看出量子隧穿不仅使得单个过程的反应速率大大提高,而且量子角切效应还能改变反应路径,从而使得经典图像下完全不发生的邻位(ortho)复合过程变得同样重要。本工作也对其他碳物质表面的氢复合过程进行了研究,发现氢在石墨烯表面的复合是最容易的。
此项工作解释了低温真空环境中关于氢分子浓度的疑问,揭示了原子核量子隧穿效应在氢复合中的关键作用。本工作中采用的研究方法有望进一步用于理解更多真实体系中的量子隧穿效应。
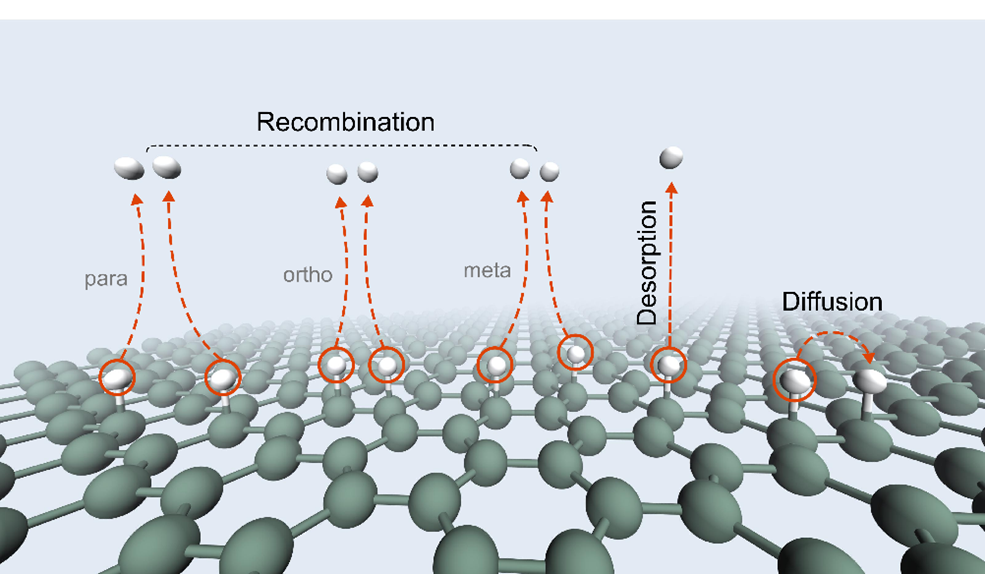
图 1. 石墨烯表面氢原子的复合(recombination),脱附(desorption)和扩散(diffusion)过程示意图。其中para,ortho和meta分别对应两个氢原子复合之前处于碳六元环的对、邻、间位。
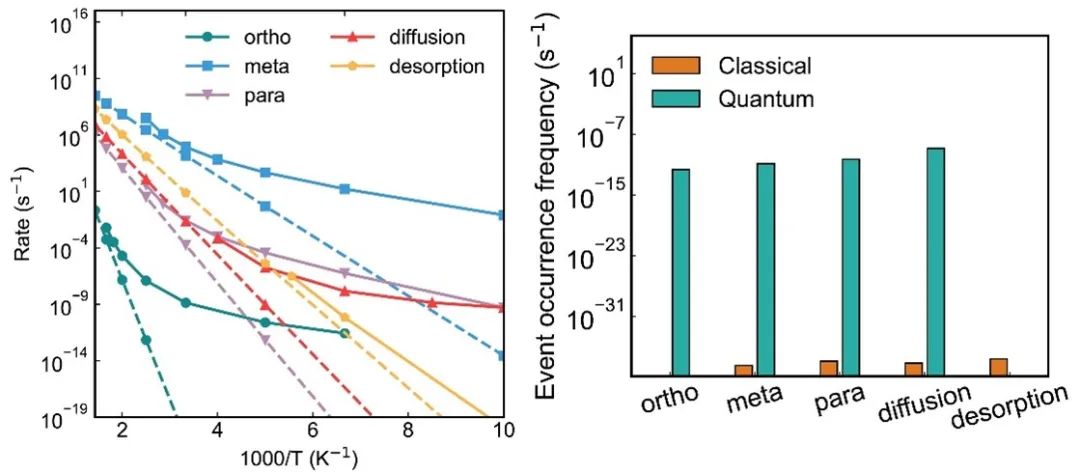
图 2. (左)经典和量子理论描述的单个过程的反应速率随着温度的变化关系。(右)基于经典和量子速率进行的动力学蒙特卡洛模拟得到的单个过程的发生频率,模拟温度为100K。
J. Phys. Chem. Lett. 2022, 13, 14, 3173–3181
Publication Date: April 1, 2022
https://doi.org/10.1021/acs.jpclett.2c00520
Copyright © 2022 American Chemical Society
本文来自ACS美国化学会,本文观点不代表石墨烯网立场,转载请联系原作者。